筋萎縮性側索硬化症とRNA結合タンパク質
2013/09/25
河原 行郎
(大阪大学大学院医学系研究科 遺伝子機能制御学)
email:河原行郎
領域融合レビュー, 2, e010 (2013) DOI: 10.7875/leading.author.2.e010
Yukio Kawahara: Amyotrophic lateral sclerosis and RNA-binding proteins.

筋萎縮性側索硬化症は上位および下位の運動ニューロンが選択的に変性し全身の筋力が低下していく神経難病である.近年,TDP-43やFUSなど,機能や構造に共通性のあるRNA結合タンパク質がその病態に深く関与していることが明らかになってきた.とくに,変性したニューロンに認められるTDP-43陽性の封入体は孤発性を含めた大部分の筋萎縮性側索硬化症に共通した病理像であり,TDP-43は発症および病態の鍵をにぎっている.また,原因遺伝子がつぎつぎと同定されるようになり,その一部は前頭側頭葉変性症とよばれる痴呆性疾患と同一の疾患スペクトラムにあることも判明してきた.現在,筋萎縮性側索硬化症においては,RNA結合タンパク質に共通する機能や標的の制御異常という観点と,これらタンパク質を中心とした凝集体の形成による毒性という観点の両面から研究が進められている.ここでは,筋萎縮性側索硬化症の研究の最新の動向について,とくに,RNA結合タンパク質やRNA代謝にかかわるタンパク質に焦点をあて解説する.
筋萎縮性側索硬化症(amyotrophic lateral sclerosis:ALS)は運動ニューロンが選択的に変性し全身の筋力が低下していく神経難病である.中年期よりのちに発症するケースが多く,いったん発症するとその進行は早く,多くは数年で呼吸不全にいたる.明らかな遺伝性を示すケースは全体の1割ほどで,多くは孤発性である.1年間の新規の発症者は人口10万人あたり約1人で,わが国には9000人弱の患者が存在する.依然として根治療法はない.2006年までは,遺伝性の筋萎縮性側索硬化症の原因として同定された遺伝子はSOD1遺伝子やALS2/Alsin遺伝子などかぎられており,また,大部分をしめる孤発性の筋萎縮性側索硬化症の発症機構において,これらの原因遺伝子がどこまでかかわっているのかという議論もたえなかった.しかし,2006年,TDP-43(TAR DNA-binding protein of 43 kDa)とよばれるRNA結合タンパク質が,変性したニューロンの細胞質の封入体においてユビキチン化,異常なリン酸化,断片化などの修飾をうけ蓄積していることが発見され1,2),これを契機に,筋萎縮性側索硬化症の研究は大きく進展した.とくに,次世代シークエンサーの実用化にともない少数の試料からでも変異遺伝子を同定できるようになったことや全エキソン解析が容易になったことにより,筋萎縮性側索硬化症の発症と関連する遺伝子変異がつぎつぎと報告されるようになった.2009年以降,毎年3~5個ほどの新規の遺伝子変異が同定されており,全体では20をこえている3).これらのなかには構造的に類似性の高いRNA結合タンパク質をコードした遺伝子が多く含まれており,RNA結合タンパク質を介したなんらかの異常が筋萎縮性側索硬化症の発症の鍵をにぎると考えられるようになってきた.
TDP-43が変性したニューロンの細胞質の封入体に蓄積しているという発見は1,2),筋萎縮性側索硬化症の研究に多くのパラダイムシフトをもたらした.
その1つ目は,同様の病理像が痴呆性疾患の一種である前頭側頭葉変性症(frontotemporal lobar degeneration:FTLD)の一部にも見い出されたことである.さらに,この発見からまもなく,頻度はきわめてまれながら,筋萎縮性側索硬化症および前頭側頭葉変性症の一部においてTDP-43をコードするTARDBP遺伝子の変異が同定された4,5)(図1).このため,筋萎縮性側索硬化症と前頭側頭葉変性症の一部はひとつの疾患スペクトラムとしてとらえられるようになり,とくに,TDP-43陽性の封入体をともなうケースはTDP-43タンパク質症(TDP-43 proteinopathy)とよばれるようになった.すなわち,筋萎縮性側索硬化症の患者には進行とともに痴呆を呈するケースがあり,また逆に,はじめ痴呆を発症しのちに筋萎縮性側索硬化症の症状を呈する場合もあることから,同じ原因遺伝子をもっていても症状の発症や進行にはばらつきのあることもわかってきた.また,これまで筋萎縮性側索硬化症においては細胞体が大きい,軸索が長いなど形態的な特異性を念頭におき運動ニューロンの選択的な脆弱性が考えられてきたが,大脳皮質のニューロンにも脆弱性という観点で類似するニューロンがあることが判明し,これらのニューロンに共通した分子特性を考慮する必要性がでてきた.
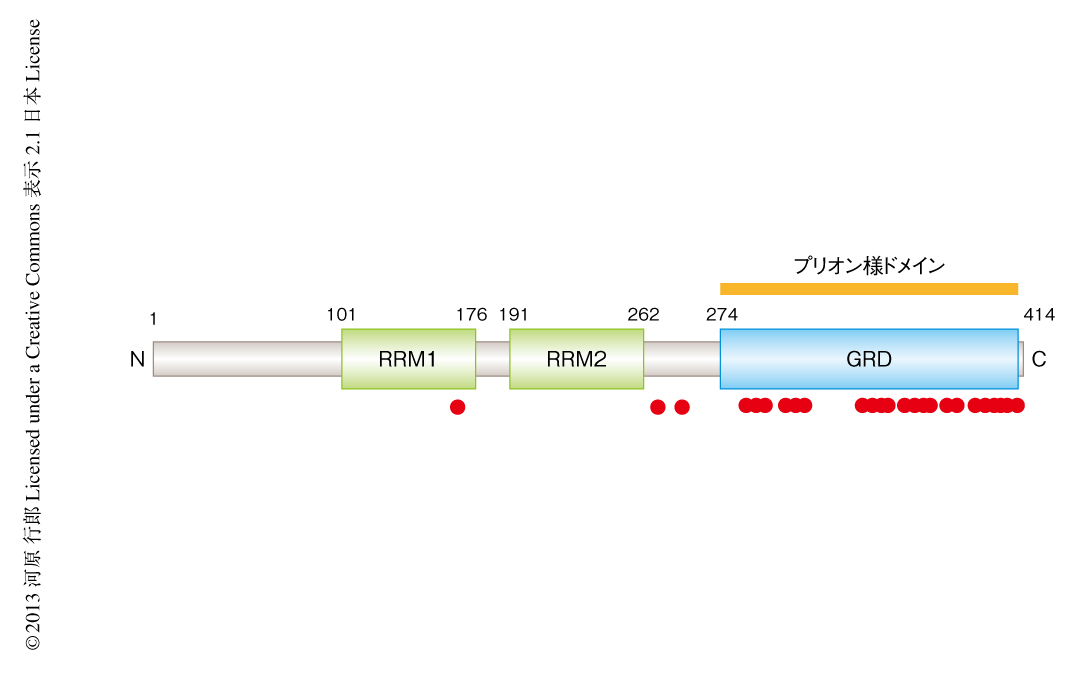
2番目は,SOD1遺伝子の変異が原因で生じる遺伝性筋萎縮性側索硬化症など一部を除き,TDP-43陽性の封入体は遺伝性あるいは孤発性にかかわらずほとんどの筋萎縮性側索硬化症に共通して認められる病理像であることが見い出された点である1,2,6).これまで,その大部分をしめる孤発性の筋萎縮性側索硬化症については病態にせまる手がかりに乏しかったが,TDP-43はほとんどの筋萎縮性側索硬化症に共通する発症に関与する鍵タンパク質として認識されるようになった.
3つ目は,本来,TDP-43は約9割が核に局在しているが,変性したニューロンにおいてはほとんどすべてが細胞質の封入体に蓄積し核から消失しているという病理所見が得られたことである1,2).したがって,変性したニューロンにおいては,本来,核ではたされているTDP-43の生理的な機能は喪失しているはずである.このため,封入体を構成するTDP-43凝集体が毒性を発揮し細胞死にいたるという毒性の獲得の面と,TDP-43の機能の異常という面から病態の研究が展開されるようになった.
次世代シークエンサーを駆使した網羅的な解析によれば,TDP-43は6000種以上のRNAの40,000以上の部位に結合しているという7,8).UG配列の豊富な領域に好んで結合し,結合部位の大部分はイントロンにある7,8).TDP-43はこれら標的となるRNAのプロセシングをさまざまなステップで制御しており,転写,スプライシング,RNA輸送,RNAの安定化にくわえ,マイクロRNAの発現の促進など,その機能は多様である9-11)(図2a).

野生型のヒトTDP-43を過剰に発現したトランスジェニックマウスは,運動ニューロンの変性により筋萎縮性側索硬化症様の症状を呈する.また,前頭側頭葉変性症において認められるような大脳皮質のニューロンの変性も認められる12).さらに,TDP-43陽性の封入体が形成され,断片化したTDP-43の蓄積も認められる.同様に,遺伝性の筋萎縮性側索硬化症において同定された変異型TDP-43を発現させたトランスジェニックマウスも,筋萎縮性側索硬化症様の症状を呈する13).これらの所見は,運動ニューロンなど特定のニューロンはTDP-43の変異の有無にかかわらず,とくに,TDP-43の過剰な状態に脆弱であることを示唆している.
TDP-43を介した細胞死の誘導の機構については,さきに述べたように,毒性の獲得と機能の異常という2つの考え方がある.まず前者については,TDP-43はC末端の領域にプリオン様ドメインを保有しており凝集しやすい性質のあることに由来する14,15)(図1).とくに,その断片化により産生されるC末端の断片は凝集の効率が高いため封入体に蓄積しやすいと考えられる(図2b).実際に,培養細胞にC末端の断片だけを発現させると凝集体を形成し細胞死が誘導される16).また,筋萎縮性側索硬化症において同定されているTARDBP遺伝子の変異のほとんどはC末端の領域に集中しており,変異が導入されるとさらに安定化し凝集しやすくなることも知られている17,18)(図1).さらに,プリオン病のように,ある細胞において凝集したTDP-43がほかの細胞にも伝搬して凝集を促進する可能性も示唆されている15).したがって,ヒトTDP-43を過剰に発現したトランスジェニックマウスのようにTDP-43が凝集しやすい状況となれば,つぎつぎと断片化と凝集体の形成による毒性が運動ニューロンに拡散しうるので,毒性獲得説では凝集体の形成にともなう細胞死の誘導が筋萎縮性側索硬化症の病態の主要な原因ではないかと考える(図2b).
一方で,TDP-43が核から細胞質の封入体へと移行すると,本来,核においてはたしている機能は喪失されることから,TDP-43によるRNA制御の異常が細胞死の主要な原因であるとする考え方もある(図2b).実際,運動ニューロンに特異的なTDP-43のノックアウトマウスは晩発性に筋萎縮性側索硬化症様の症状を呈する19).また,TDP-43が細胞質の封入体に移行するまえの段階からRNA制御に異常をきたしていることを示唆する所見もあることから10,20),凝集体の形成は細胞死を加速させるものの,初期的なニューロンの変性の原因はTDP-43の機能の異常にある可能性が示唆されている.いずれにしても現状では,なぜ運動ニューロンなど特定のニューロンがTDP-43の過剰な状態に脆弱であるのか,また,筋萎縮性側索硬化症の患者の運動ニューロンにおいてTDP-43の代謝に初期的にどのような異常が生じているのかは解決されていない.かりに,TDP-43の機能の異常が初期的なニューロンの変性の原因であるなら,特異的なTDP-43の機能あるいはその標的が運動ニューロンに存在すると予想されることから,これらが同定されれば病態の解明にむけ大きく前進すると考えられる.
2009年,遺伝性筋萎縮性側索硬化症の原因遺伝子としてFUS(fused in sarcoma)遺伝子が同定された21,22)(図3).この遺伝子の産物FUSはTDP-43と同様にRNA結合タンパク質であること,前頭側頭葉変性症の一部にFUS遺伝子の変異が同定されたことから,筋萎縮性側索硬化症および前頭側頭葉変性症の病態には共通してRNA結合タンパク質が関与するのではないかと考えられるようになった.FUSは核により多く局在するが,TDP-43と比べると細胞質に局在する割合は多い.また,5500種以上のRNAの44,000以上の部位に結合することが知られており,スプライシングやmRNAの安定化などRNAの機能に多様に関与している23-25).しかしながら,おおまかにはTDP-43とは標的や結合部位は異なっており,共通する標的はかぎられている23,24).筋萎縮性側索硬化症において同定されている変異はN末端側のプリオン様ドメインとC末端側の核局在化シグナルの付近に多い26)(図3).核局在化シグナルの側に生じた変異はFUSの局在を核から細胞質へと変えることもあり,これらの変異をもつ患者はより早期に発症する傾向にある27).また,TDP-43と同様に,プリオン様ドメインがあるため凝集体を形成しやすく,FUS遺伝子の変異をともなう筋萎縮性側索硬化症では多くのケースにおいてFUS陽性の封入体が認められる21,22).一方で,FUS陽性の封入体をもつケースではTDP-43の異常をともなわないこと,FUS遺伝子に変異をもたない前頭側頭葉変性症でもFUS陽性の封入体の認められるケースのあることから28),筋萎縮性側索硬化症および前頭側頭葉変性症にはTDP-43に依存しないFUSを介した発症の病態があると考えられている.実際,野生型のヒトFUSを過剰に発現するトランスジェニックマウスは麻痺を生じ生後3カ月で死にいたる29).また,変異型のヒトFUSの発現を生後から誘導したラットでは生後70日で麻痺を生じ死にいたる30).このように,TDP-43と同様に,モデル動物においては変異の有無にかかわらずFUSの過剰発現により運動ニューロンに症状を呈する.
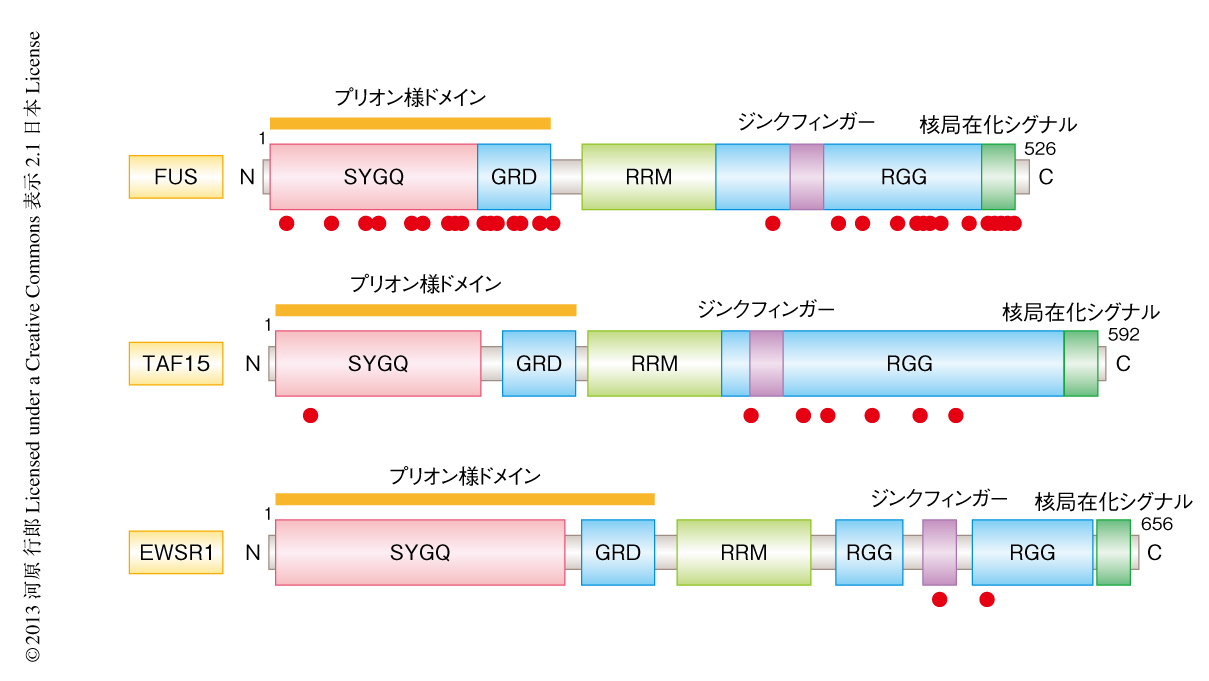
FUSは構造的に近いTAF15(TATA-binding protein associated factor 15)とEWSR1(Ewing sarcoma breakpoint region 1)とともにFETファミリーを構成する(図3).これらは構造的に近いだけでなく,さまざまながんにおいて染色体転座の生じやすい領域にあるという点もあわせもつ26).このため,TAF15遺伝子およびEWSR1遺伝子についても変異解析が進められ,筋萎縮性側索硬化症において特異的な変異が同定された31,32)(図3).また,FETファミリーには共通してプリオン様ドメインが存在するため凝集しやすい傾向にあり,本来はおもに核に局在しているFETファミリータンパク質は変性したニューロンにおいては細胞質に移行している.このように,TDP-43とFETファミリーについては病理像においては相互に排他的であるが,構造や生理的な機能においては共通点が多く,筋萎縮性側索硬化症の発症にいたる経路はある程度まで類似しているのではないかと予想される.これに関しても,TDP-43とFETファミリーとのあいだには共通したRNA制御の機能や標的RNAがあり,その制御の異常により筋萎縮性側索硬化症は発症するのではないかという考え方と,構造的な類似性からともに凝集しやすく,これにより毒性を獲得するのではないかという考え方がある.
2010年から2011年にかけて,筋萎縮性側索硬化症の病態を考えるうえで,さらなる重要な発見があいついだ.これまで,遺伝性脊髄小脳変性症や筋緊張性ジストロフィーなどにおいては特定のリピート配列の異常な伸長が原因であることが知られ,リピート病とよばれてきた.とくに,Huntington病などではタンパク質コード領域に生じるCAGリピート配列の異常な伸長がポリグルタミン鎖へと翻訳されることから,ポリグルタミン病と総称される.筋萎縮性側索硬化症はこういったリピート配列の伸長とは無縁と考えられてきたが,2010年,ポリグルタミン病の一種である遺伝性脊髄小脳変性症2型の原因となるATXN2遺伝子(Ataxin-2をコードする)のCAGリピート配列が,筋萎縮性側索硬化症の患者において有意に伸長していることが報告された33).健常者はこのリピートの回数が23回以下であるのに対し,遺伝性脊髄小脳変性症2型の患者では34回以上に伸長している.一方で,筋萎縮性側索硬化症の患者ではこの中間の長さ,27~33回のリピート数を保有しているケースが有意に多かった.そののち,Huntington病や遺伝性脊髄小脳変性症1型などほかのリピート病の原因となる遺伝子のリピートの回数と筋萎縮性側索硬化症の発症との相関も解析されたが,有意な相関はATXN2遺伝子のみで認められた34).この結果から,Ataxin-2が筋萎縮性側索硬化症の発症に関与していると考えられるようになったが,現状では,CAGリピート配列の中等度の伸長だけで筋萎縮性側索硬化症が発症するかどうかは不明であり,発症を高める危険因子として認識されている.Ataxin-2はポリA鎖結合タンパク質であるPABPC1と直接に結合することからなんらかのRNA代謝に関与していると考えられており,また,TDP-43とAtaxin-2はRNAを介し結合している33,35).ショウジョウバエの眼にTDP-43を過剰に発現させると神経変性が生じるが,ここにさらにAtaxin-2を過剰に発現させると神経変性は加速することから,Ataxin-2はTDP-43を介した神経変性の分子機構を促進する機能があると推測されている33).
2011年,C9orf72遺伝子のイントロンにおけるGGGGCCリピート配列の異常な伸長が一部の筋萎縮性側索硬化症および前頭側頭葉変性症の原因として同定された36,37).この遺伝子変異は欧米において比較的多く,遺伝性筋萎縮性側索硬化症の30~40%,孤発性筋萎縮性側索硬化症の5~8%に認められたが,わが国ではきわめて少数例であったことから強い創始者効果が想定された.一方,日本の紀伊半島には筋萎縮性側索硬化症の多発する地域のあることが知られているが,その一部はC9orf72遺伝子のGGGGCCリピート配列の異常な伸長をもつことが確認された38).長年にわたり,筋萎縮性側索硬化症の多発する理由についてはさまざまな仮説が提唱されてきた経緯もあり,その原因の一端が明らかになった意義は大きい.
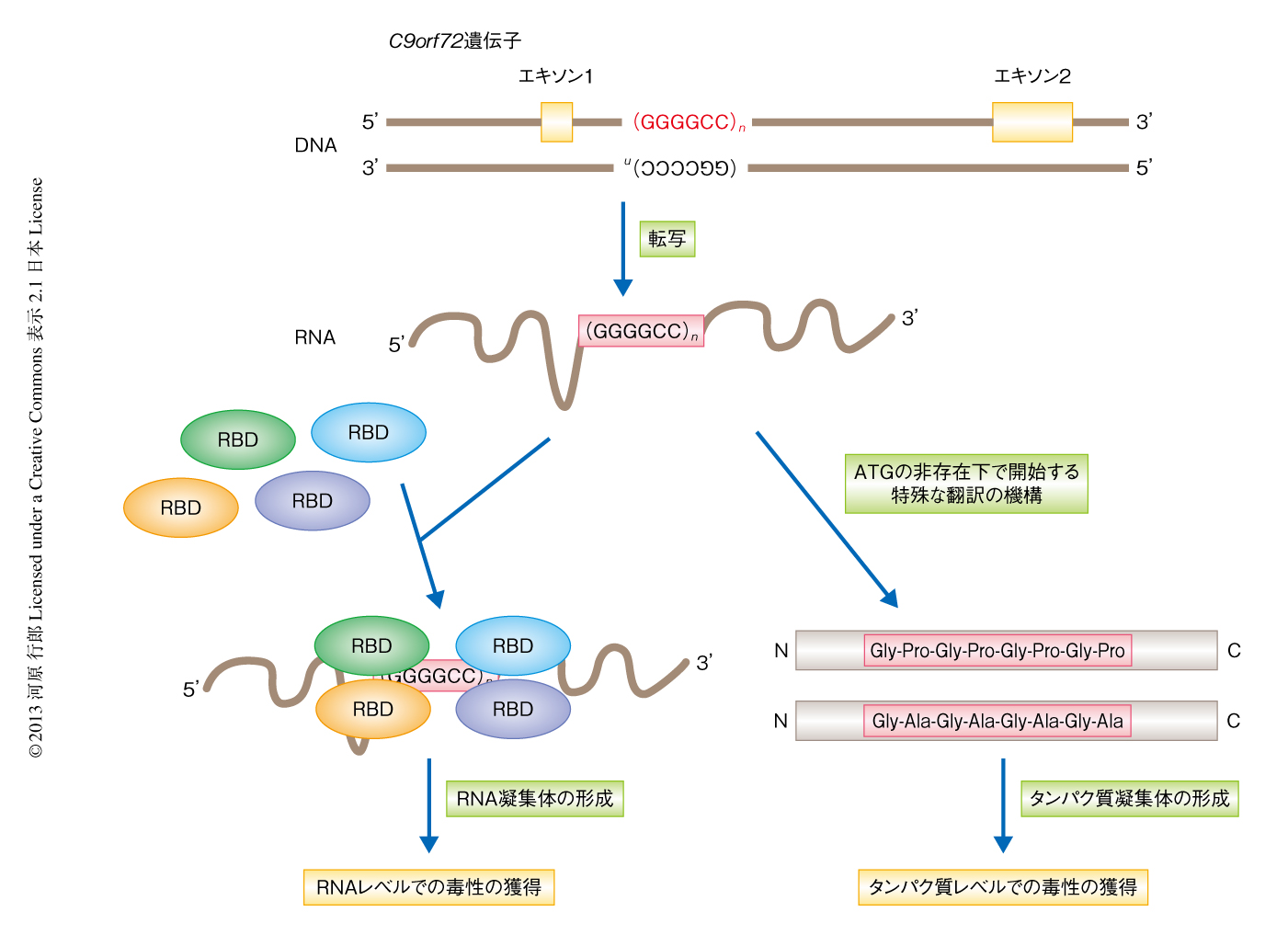
この発見により,筋萎縮性側索硬化症と前頭側頭葉変性症とが同じ疾患スペクトラムにあることを再認識することになっただけでなく,筋萎縮性側索硬化症および前頭側頭葉変性症がほかのリピート病と共通した発症機構をもつ可能性も考えられるようになった.リピート病では一般に,リピート配列から翻訳されたタンパク質が毒性を発揮する場合と,リピート配列を含むRNAそのものが毒性を発揮する場合とが知られているが,近年では,いずれのリピート病においてもその両者が複合的に関与していると考えられるようになってきている.実際に,筋萎縮性側索硬化症および前頭側頭葉変性症においては,当初,C9orf72遺伝子のGGGGCCリピート配列はイントロンにあることからこのリピート配列を含むRNAが毒性を発揮すると考えられ,さまざまなRNA結合タンパク質を吸着して形成される細胞内構造体であるRNA凝集体(RNA foci)の観察されることが報告された36,37)(図4).しかしそののち,リピート配列に関連し開始コドンであるATGの非存在下にて開始する特殊な形式での翻訳(repeat-associated non-ATG-initiated translation,RAN翻訳)機構を介して,C9orf72遺伝子のGGGGCCリピート配列をもつ領域からタンパク質が発現していることが発見された39)(新着論文レビュー でも掲載)(図4).C9orf72遺伝子のGGGGCCリピート配列が異常に伸長している患者の脳においては,TDP-43陽性の封入体とともにTDP-43陰性の封入体も認められるが,このTDP-43陰性の封入体にはGGGGCCリピート配列から翻訳されたタンパク質が凝集していることも明らかになった39).今後は,C9orf72遺伝子のGGGGCCリピート配列の伸長がどのようにTDP-43の機能や局在に影響するのか,その分子機構が明らかになれば,筋萎縮性側索硬化症の全般に共通するTDP-43を介した発症機構の上流にせまることができると期待される.
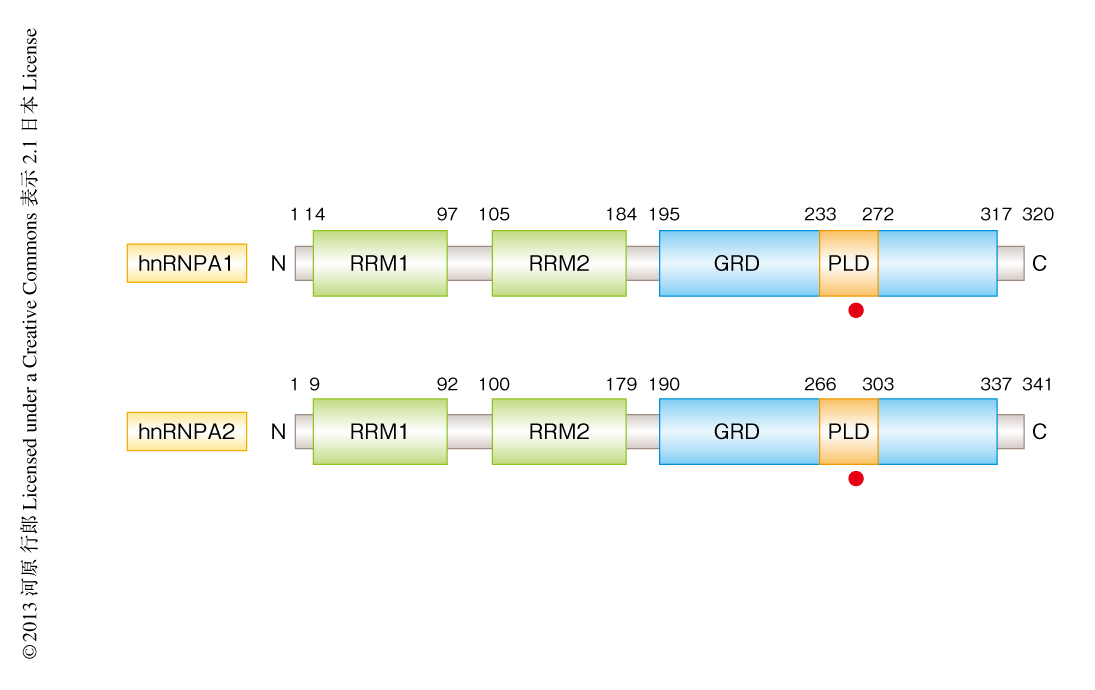
最近になり,頻度はきわめてまれではあるが筋萎縮性側索硬化症において新たなRNA結合タンパク質の変異が同定された40)(図5).もともと,封入体ミオパチーおよび骨Paget病に前頭側頭葉変性症や筋萎縮性側索硬化症の合併する,常染色体優性遺伝性のまれな疾患があった.この疾患では病変部位にTDP-43陽性の封入体が観察され,近年,多系統タンパク質症(multisystem proteinopathy:MSP)とよばれるようになっている.その原因遺伝子のひとつとしてVCP(Valosin-containing protein)遺伝子が同定されていたが41),このVCP遺伝子の変異はそののち,非複合型筋萎縮性側索硬化症,前頭側頭葉変性症,封入体ミオパチー,骨Paget病の患者からも同定された.さらに最近,多系統タンパク質症の新たな遺伝子変異がRNA結合タンパク質をコードするhnRNPA2B1遺伝子およびhnRNPA1遺伝子において同定された40)(図5).また,多系統タンパク質症だけでなく,筋萎縮性側索硬化症においてもhnRNPA1遺伝子の変異が同定されるにいたった40).これらの事実は,同じ遺伝子に起因する異常が,筋萎縮性側索硬化症や前頭側頭葉変性症など神経変性疾患にとどまらず,筋や骨の疾患の病態にも共通している可能性を示唆しており,疾患スペクトラムをさらに広げて考える必要性がでてきた.これらの疾患においては,本来は核に局在し一部はTDP-43と生理的に結合しているhnRNPA1やhnRNPA2B1が細胞質の封入体に蓄積しており,この病理像はVCP遺伝子の変異に起因するケースでも認められる.hnRNPA1およびhnRNPA2B1はTDP-43やFETファミリーと同じくプリオン様ドメインをもっており,このプリオン様ドメインにおける変異により凝集しやすくなる(図5).また,FUSの局在は正常である一方,TDP-43陽性の封入体も観察されるが,この封入体におけるhnRNPA1やhnRNPA2B1との共局在は一部にとどまる40).このため,C9orf72遺伝子のGGGGCCリピート配列のケースと同様に,hnRNPA1やhnRNPA2B1における変異がどのようにTDP-43の機能や局在に影響するのか,今後,その分子機構が明らかとなることを期待したい.
これまで,筋萎縮性側索硬化症においては病態に関連するタンパク質がほとんど未知であったことから,仮説主導型の研究が主流であった.しかし,TDP-43の発見ののち多くの知見が得られるようになり,筋萎縮性側索硬化症という疾患の単位を再考する必要性にせまられるほど,さまざまな疾患と発症の病態が共通している可能性が示唆されるようになってきた.また,TDP-43を中心として,それより上流に位置するタンパク質や下流にある機構もしだいに解明されつつある.今回は,これら筋萎縮性側索硬化症に関連するすべての遺伝子やタンパク質はとりあげなかったが,このなかには,ADAR2,Senataxin,AngiogeninなどRNA制御に関連するタンパク質や,Ubiquilin-2,p62(SQSTM1)などユビキチン-プロテアソーム系に関連するタンパク質が含まれている.いずれにしても,大部分をしめる孤発性の筋萎縮性側索硬化症の発症機構を解明するには,TDP-43の代謝がどのような分子機構により破綻するのかを解明することが鍵をにぎっている.近い将来,これらの問題が解決され治療法および予防法が確立できるよう,筆者自身も尽力したい.
略歴:2004年 東京大学大学院医学系研究科博士課程 修了,同年 米国Wistar Instituteポスドク研究員,2009年 大阪大学大学院医学系研究科 テニュアトラック准教授を経て,2013年 同 独立准教授.
研究テーマ:RNAの機能や修飾の異常を切り口に,筋萎縮性側索硬化症など疾患の病態を解明する“RNA病態学”をメインテーマにあげている.
研究室URL:http://www.med.osaka-u.ac.jp/pub/rna/index.html
© 2013 河原 行郎 Licensed under CC 表示 2.1 日本
(大阪大学大学院医学系研究科 遺伝子機能制御学)
email:河原行郎
領域融合レビュー, 2, e010 (2013) DOI: 10.7875/leading.author.2.e010
Yukio Kawahara: Amyotrophic lateral sclerosis and RNA-binding proteins.
要 約
筋萎縮性側索硬化症は上位および下位の運動ニューロンが選択的に変性し全身の筋力が低下していく神経難病である.近年,TDP-43やFUSなど,機能や構造に共通性のあるRNA結合タンパク質がその病態に深く関与していることが明らかになってきた.とくに,変性したニューロンに認められるTDP-43陽性の封入体は孤発性を含めた大部分の筋萎縮性側索硬化症に共通した病理像であり,TDP-43は発症および病態の鍵をにぎっている.また,原因遺伝子がつぎつぎと同定されるようになり,その一部は前頭側頭葉変性症とよばれる痴呆性疾患と同一の疾患スペクトラムにあることも判明してきた.現在,筋萎縮性側索硬化症においては,RNA結合タンパク質に共通する機能や標的の制御異常という観点と,これらタンパク質を中心とした凝集体の形成による毒性という観点の両面から研究が進められている.ここでは,筋萎縮性側索硬化症の研究の最新の動向について,とくに,RNA結合タンパク質やRNA代謝にかかわるタンパク質に焦点をあて解説する.
はじめに
筋萎縮性側索硬化症(amyotrophic lateral sclerosis:ALS)は運動ニューロンが選択的に変性し全身の筋力が低下していく神経難病である.中年期よりのちに発症するケースが多く,いったん発症するとその進行は早く,多くは数年で呼吸不全にいたる.明らかな遺伝性を示すケースは全体の1割ほどで,多くは孤発性である.1年間の新規の発症者は人口10万人あたり約1人で,わが国には9000人弱の患者が存在する.依然として根治療法はない.2006年までは,遺伝性の筋萎縮性側索硬化症の原因として同定された遺伝子はSOD1遺伝子やALS2/Alsin遺伝子などかぎられており,また,大部分をしめる孤発性の筋萎縮性側索硬化症の発症機構において,これらの原因遺伝子がどこまでかかわっているのかという議論もたえなかった.しかし,2006年,TDP-43(TAR DNA-binding protein of 43 kDa)とよばれるRNA結合タンパク質が,変性したニューロンの細胞質の封入体においてユビキチン化,異常なリン酸化,断片化などの修飾をうけ蓄積していることが発見され1,2),これを契機に,筋萎縮性側索硬化症の研究は大きく進展した.とくに,次世代シークエンサーの実用化にともない少数の試料からでも変異遺伝子を同定できるようになったことや全エキソン解析が容易になったことにより,筋萎縮性側索硬化症の発症と関連する遺伝子変異がつぎつぎと報告されるようになった.2009年以降,毎年3~5個ほどの新規の遺伝子変異が同定されており,全体では20をこえている3).これらのなかには構造的に類似性の高いRNA結合タンパク質をコードした遺伝子が多く含まれており,RNA結合タンパク質を介したなんらかの異常が筋萎縮性側索硬化症の発症の鍵をにぎると考えられるようになってきた.
1.筋萎縮性側索硬化症とTDP-43
TDP-43が変性したニューロンの細胞質の封入体に蓄積しているという発見は1,2),筋萎縮性側索硬化症の研究に多くのパラダイムシフトをもたらした.
その1つ目は,同様の病理像が痴呆性疾患の一種である前頭側頭葉変性症(frontotemporal lobar degeneration:FTLD)の一部にも見い出されたことである.さらに,この発見からまもなく,頻度はきわめてまれながら,筋萎縮性側索硬化症および前頭側頭葉変性症の一部においてTDP-43をコードするTARDBP遺伝子の変異が同定された4,5)(図1).このため,筋萎縮性側索硬化症と前頭側頭葉変性症の一部はひとつの疾患スペクトラムとしてとらえられるようになり,とくに,TDP-43陽性の封入体をともなうケースはTDP-43タンパク質症(TDP-43 proteinopathy)とよばれるようになった.すなわち,筋萎縮性側索硬化症の患者には進行とともに痴呆を呈するケースがあり,また逆に,はじめ痴呆を発症しのちに筋萎縮性側索硬化症の症状を呈する場合もあることから,同じ原因遺伝子をもっていても症状の発症や進行にはばらつきのあることもわかってきた.また,これまで筋萎縮性側索硬化症においては細胞体が大きい,軸索が長いなど形態的な特異性を念頭におき運動ニューロンの選択的な脆弱性が考えられてきたが,大脳皮質のニューロンにも脆弱性という観点で類似するニューロンがあることが判明し,これらのニューロンに共通した分子特性を考慮する必要性がでてきた.
2番目は,SOD1遺伝子の変異が原因で生じる遺伝性筋萎縮性側索硬化症など一部を除き,TDP-43陽性の封入体は遺伝性あるいは孤発性にかかわらずほとんどの筋萎縮性側索硬化症に共通して認められる病理像であることが見い出された点である1,2,6).これまで,その大部分をしめる孤発性の筋萎縮性側索硬化症については病態にせまる手がかりに乏しかったが,TDP-43はほとんどの筋萎縮性側索硬化症に共通する発症に関与する鍵タンパク質として認識されるようになった.
3つ目は,本来,TDP-43は約9割が核に局在しているが,変性したニューロンにおいてはほとんどすべてが細胞質の封入体に蓄積し核から消失しているという病理所見が得られたことである1,2).したがって,変性したニューロンにおいては,本来,核ではたされているTDP-43の生理的な機能は喪失しているはずである.このため,封入体を構成するTDP-43凝集体が毒性を発揮し細胞死にいたるという毒性の獲得の面と,TDP-43の機能の異常という面から病態の研究が展開されるようになった.
2.TDP-43の機能と病態
次世代シークエンサーを駆使した網羅的な解析によれば,TDP-43は6000種以上のRNAの40,000以上の部位に結合しているという7,8).UG配列の豊富な領域に好んで結合し,結合部位の大部分はイントロンにある7,8).TDP-43はこれら標的となるRNAのプロセシングをさまざまなステップで制御しており,転写,スプライシング,RNA輸送,RNAの安定化にくわえ,マイクロRNAの発現の促進など,その機能は多様である9-11)(図2a).
野生型のヒトTDP-43を過剰に発現したトランスジェニックマウスは,運動ニューロンの変性により筋萎縮性側索硬化症様の症状を呈する.また,前頭側頭葉変性症において認められるような大脳皮質のニューロンの変性も認められる12).さらに,TDP-43陽性の封入体が形成され,断片化したTDP-43の蓄積も認められる.同様に,遺伝性の筋萎縮性側索硬化症において同定された変異型TDP-43を発現させたトランスジェニックマウスも,筋萎縮性側索硬化症様の症状を呈する13).これらの所見は,運動ニューロンなど特定のニューロンはTDP-43の変異の有無にかかわらず,とくに,TDP-43の過剰な状態に脆弱であることを示唆している.
TDP-43を介した細胞死の誘導の機構については,さきに述べたように,毒性の獲得と機能の異常という2つの考え方がある.まず前者については,TDP-43はC末端の領域にプリオン様ドメインを保有しており凝集しやすい性質のあることに由来する14,15)(図1).とくに,その断片化により産生されるC末端の断片は凝集の効率が高いため封入体に蓄積しやすいと考えられる(図2b).実際に,培養細胞にC末端の断片だけを発現させると凝集体を形成し細胞死が誘導される16).また,筋萎縮性側索硬化症において同定されているTARDBP遺伝子の変異のほとんどはC末端の領域に集中しており,変異が導入されるとさらに安定化し凝集しやすくなることも知られている17,18)(図1).さらに,プリオン病のように,ある細胞において凝集したTDP-43がほかの細胞にも伝搬して凝集を促進する可能性も示唆されている15).したがって,ヒトTDP-43を過剰に発現したトランスジェニックマウスのようにTDP-43が凝集しやすい状況となれば,つぎつぎと断片化と凝集体の形成による毒性が運動ニューロンに拡散しうるので,毒性獲得説では凝集体の形成にともなう細胞死の誘導が筋萎縮性側索硬化症の病態の主要な原因ではないかと考える(図2b).
一方で,TDP-43が核から細胞質の封入体へと移行すると,本来,核においてはたしている機能は喪失されることから,TDP-43によるRNA制御の異常が細胞死の主要な原因であるとする考え方もある(図2b).実際,運動ニューロンに特異的なTDP-43のノックアウトマウスは晩発性に筋萎縮性側索硬化症様の症状を呈する19).また,TDP-43が細胞質の封入体に移行するまえの段階からRNA制御に異常をきたしていることを示唆する所見もあることから10,20),凝集体の形成は細胞死を加速させるものの,初期的なニューロンの変性の原因はTDP-43の機能の異常にある可能性が示唆されている.いずれにしても現状では,なぜ運動ニューロンなど特定のニューロンがTDP-43の過剰な状態に脆弱であるのか,また,筋萎縮性側索硬化症の患者の運動ニューロンにおいてTDP-43の代謝に初期的にどのような異常が生じているのかは解決されていない.かりに,TDP-43の機能の異常が初期的なニューロンの変性の原因であるなら,特異的なTDP-43の機能あるいはその標的が運動ニューロンに存在すると予想されることから,これらが同定されれば病態の解明にむけ大きく前進すると考えられる.
3.筋萎縮性側索硬化症とFETファミリー
2009年,遺伝性筋萎縮性側索硬化症の原因遺伝子としてFUS(fused in sarcoma)遺伝子が同定された21,22)(図3).この遺伝子の産物FUSはTDP-43と同様にRNA結合タンパク質であること,前頭側頭葉変性症の一部にFUS遺伝子の変異が同定されたことから,筋萎縮性側索硬化症および前頭側頭葉変性症の病態には共通してRNA結合タンパク質が関与するのではないかと考えられるようになった.FUSは核により多く局在するが,TDP-43と比べると細胞質に局在する割合は多い.また,5500種以上のRNAの44,000以上の部位に結合することが知られており,スプライシングやmRNAの安定化などRNAの機能に多様に関与している23-25).しかしながら,おおまかにはTDP-43とは標的や結合部位は異なっており,共通する標的はかぎられている23,24).筋萎縮性側索硬化症において同定されている変異はN末端側のプリオン様ドメインとC末端側の核局在化シグナルの付近に多い26)(図3).核局在化シグナルの側に生じた変異はFUSの局在を核から細胞質へと変えることもあり,これらの変異をもつ患者はより早期に発症する傾向にある27).また,TDP-43と同様に,プリオン様ドメインがあるため凝集体を形成しやすく,FUS遺伝子の変異をともなう筋萎縮性側索硬化症では多くのケースにおいてFUS陽性の封入体が認められる21,22).一方で,FUS陽性の封入体をもつケースではTDP-43の異常をともなわないこと,FUS遺伝子に変異をもたない前頭側頭葉変性症でもFUS陽性の封入体の認められるケースのあることから28),筋萎縮性側索硬化症および前頭側頭葉変性症にはTDP-43に依存しないFUSを介した発症の病態があると考えられている.実際,野生型のヒトFUSを過剰に発現するトランスジェニックマウスは麻痺を生じ生後3カ月で死にいたる29).また,変異型のヒトFUSの発現を生後から誘導したラットでは生後70日で麻痺を生じ死にいたる30).このように,TDP-43と同様に,モデル動物においては変異の有無にかかわらずFUSの過剰発現により運動ニューロンに症状を呈する.
FUSは構造的に近いTAF15(TATA-binding protein associated factor 15)とEWSR1(Ewing sarcoma breakpoint region 1)とともにFETファミリーを構成する(図3).これらは構造的に近いだけでなく,さまざまながんにおいて染色体転座の生じやすい領域にあるという点もあわせもつ26).このため,TAF15遺伝子およびEWSR1遺伝子についても変異解析が進められ,筋萎縮性側索硬化症において特異的な変異が同定された31,32)(図3).また,FETファミリーには共通してプリオン様ドメインが存在するため凝集しやすい傾向にあり,本来はおもに核に局在しているFETファミリータンパク質は変性したニューロンにおいては細胞質に移行している.このように,TDP-43とFETファミリーについては病理像においては相互に排他的であるが,構造や生理的な機能においては共通点が多く,筋萎縮性側索硬化症の発症にいたる経路はある程度まで類似しているのではないかと予想される.これに関しても,TDP-43とFETファミリーとのあいだには共通したRNA制御の機能や標的RNAがあり,その制御の異常により筋萎縮性側索硬化症は発症するのではないかという考え方と,構造的な類似性からともに凝集しやすく,これにより毒性を獲得するのではないかという考え方がある.
4.筋萎縮性側索硬化症とリピート配列の異常な伸長
2010年から2011年にかけて,筋萎縮性側索硬化症の病態を考えるうえで,さらなる重要な発見があいついだ.これまで,遺伝性脊髄小脳変性症や筋緊張性ジストロフィーなどにおいては特定のリピート配列の異常な伸長が原因であることが知られ,リピート病とよばれてきた.とくに,Huntington病などではタンパク質コード領域に生じるCAGリピート配列の異常な伸長がポリグルタミン鎖へと翻訳されることから,ポリグルタミン病と総称される.筋萎縮性側索硬化症はこういったリピート配列の伸長とは無縁と考えられてきたが,2010年,ポリグルタミン病の一種である遺伝性脊髄小脳変性症2型の原因となるATXN2遺伝子(Ataxin-2をコードする)のCAGリピート配列が,筋萎縮性側索硬化症の患者において有意に伸長していることが報告された33).健常者はこのリピートの回数が23回以下であるのに対し,遺伝性脊髄小脳変性症2型の患者では34回以上に伸長している.一方で,筋萎縮性側索硬化症の患者ではこの中間の長さ,27~33回のリピート数を保有しているケースが有意に多かった.そののち,Huntington病や遺伝性脊髄小脳変性症1型などほかのリピート病の原因となる遺伝子のリピートの回数と筋萎縮性側索硬化症の発症との相関も解析されたが,有意な相関はATXN2遺伝子のみで認められた34).この結果から,Ataxin-2が筋萎縮性側索硬化症の発症に関与していると考えられるようになったが,現状では,CAGリピート配列の中等度の伸長だけで筋萎縮性側索硬化症が発症するかどうかは不明であり,発症を高める危険因子として認識されている.Ataxin-2はポリA鎖結合タンパク質であるPABPC1と直接に結合することからなんらかのRNA代謝に関与していると考えられており,また,TDP-43とAtaxin-2はRNAを介し結合している33,35).ショウジョウバエの眼にTDP-43を過剰に発現させると神経変性が生じるが,ここにさらにAtaxin-2を過剰に発現させると神経変性は加速することから,Ataxin-2はTDP-43を介した神経変性の分子機構を促進する機能があると推測されている33).
2011年,C9orf72遺伝子のイントロンにおけるGGGGCCリピート配列の異常な伸長が一部の筋萎縮性側索硬化症および前頭側頭葉変性症の原因として同定された36,37).この遺伝子変異は欧米において比較的多く,遺伝性筋萎縮性側索硬化症の30~40%,孤発性筋萎縮性側索硬化症の5~8%に認められたが,わが国ではきわめて少数例であったことから強い創始者効果が想定された.一方,日本の紀伊半島には筋萎縮性側索硬化症の多発する地域のあることが知られているが,その一部はC9orf72遺伝子のGGGGCCリピート配列の異常な伸長をもつことが確認された38).長年にわたり,筋萎縮性側索硬化症の多発する理由についてはさまざまな仮説が提唱されてきた経緯もあり,その原因の一端が明らかになった意義は大きい.
この発見により,筋萎縮性側索硬化症と前頭側頭葉変性症とが同じ疾患スペクトラムにあることを再認識することになっただけでなく,筋萎縮性側索硬化症および前頭側頭葉変性症がほかのリピート病と共通した発症機構をもつ可能性も考えられるようになった.リピート病では一般に,リピート配列から翻訳されたタンパク質が毒性を発揮する場合と,リピート配列を含むRNAそのものが毒性を発揮する場合とが知られているが,近年では,いずれのリピート病においてもその両者が複合的に関与していると考えられるようになってきている.実際に,筋萎縮性側索硬化症および前頭側頭葉変性症においては,当初,C9orf72遺伝子のGGGGCCリピート配列はイントロンにあることからこのリピート配列を含むRNAが毒性を発揮すると考えられ,さまざまなRNA結合タンパク質を吸着して形成される細胞内構造体であるRNA凝集体(RNA foci)の観察されることが報告された36,37)(図4).しかしそののち,リピート配列に関連し開始コドンであるATGの非存在下にて開始する特殊な形式での翻訳(repeat-associated non-ATG-initiated translation,RAN翻訳)機構を介して,C9orf72遺伝子のGGGGCCリピート配列をもつ領域からタンパク質が発現していることが発見された39)(新着論文レビュー でも掲載)(図4).C9orf72遺伝子のGGGGCCリピート配列が異常に伸長している患者の脳においては,TDP-43陽性の封入体とともにTDP-43陰性の封入体も認められるが,このTDP-43陰性の封入体にはGGGGCCリピート配列から翻訳されたタンパク質が凝集していることも明らかになった39).今後は,C9orf72遺伝子のGGGGCCリピート配列の伸長がどのようにTDP-43の機能や局在に影響するのか,その分子機構が明らかになれば,筋萎縮性側索硬化症の全般に共通するTDP-43を介した発症機構の上流にせまることができると期待される.
5.筋萎縮性側索硬化症と多系統タンパク質症
最近になり,頻度はきわめてまれではあるが筋萎縮性側索硬化症において新たなRNA結合タンパク質の変異が同定された40)(図5).もともと,封入体ミオパチーおよび骨Paget病に前頭側頭葉変性症や筋萎縮性側索硬化症の合併する,常染色体優性遺伝性のまれな疾患があった.この疾患では病変部位にTDP-43陽性の封入体が観察され,近年,多系統タンパク質症(multisystem proteinopathy:MSP)とよばれるようになっている.その原因遺伝子のひとつとしてVCP(Valosin-containing protein)遺伝子が同定されていたが41),このVCP遺伝子の変異はそののち,非複合型筋萎縮性側索硬化症,前頭側頭葉変性症,封入体ミオパチー,骨Paget病の患者からも同定された.さらに最近,多系統タンパク質症の新たな遺伝子変異がRNA結合タンパク質をコードするhnRNPA2B1遺伝子およびhnRNPA1遺伝子において同定された40)(図5).また,多系統タンパク質症だけでなく,筋萎縮性側索硬化症においてもhnRNPA1遺伝子の変異が同定されるにいたった40).これらの事実は,同じ遺伝子に起因する異常が,筋萎縮性側索硬化症や前頭側頭葉変性症など神経変性疾患にとどまらず,筋や骨の疾患の病態にも共通している可能性を示唆しており,疾患スペクトラムをさらに広げて考える必要性がでてきた.これらの疾患においては,本来は核に局在し一部はTDP-43と生理的に結合しているhnRNPA1やhnRNPA2B1が細胞質の封入体に蓄積しており,この病理像はVCP遺伝子の変異に起因するケースでも認められる.hnRNPA1およびhnRNPA2B1はTDP-43やFETファミリーと同じくプリオン様ドメインをもっており,このプリオン様ドメインにおける変異により凝集しやすくなる(図5).また,FUSの局在は正常である一方,TDP-43陽性の封入体も観察されるが,この封入体におけるhnRNPA1やhnRNPA2B1との共局在は一部にとどまる40).このため,C9orf72遺伝子のGGGGCCリピート配列のケースと同様に,hnRNPA1やhnRNPA2B1における変異がどのようにTDP-43の機能や局在に影響するのか,今後,その分子機構が明らかとなることを期待したい.
おわりに
これまで,筋萎縮性側索硬化症においては病態に関連するタンパク質がほとんど未知であったことから,仮説主導型の研究が主流であった.しかし,TDP-43の発見ののち多くの知見が得られるようになり,筋萎縮性側索硬化症という疾患の単位を再考する必要性にせまられるほど,さまざまな疾患と発症の病態が共通している可能性が示唆されるようになってきた.また,TDP-43を中心として,それより上流に位置するタンパク質や下流にある機構もしだいに解明されつつある.今回は,これら筋萎縮性側索硬化症に関連するすべての遺伝子やタンパク質はとりあげなかったが,このなかには,ADAR2,Senataxin,AngiogeninなどRNA制御に関連するタンパク質や,Ubiquilin-2,p62(SQSTM1)などユビキチン-プロテアソーム系に関連するタンパク質が含まれている.いずれにしても,大部分をしめる孤発性の筋萎縮性側索硬化症の発症機構を解明するには,TDP-43の代謝がどのような分子機構により破綻するのかを解明することが鍵をにぎっている.近い将来,これらの問題が解決され治療法および予防法が確立できるよう,筆者自身も尽力したい.
文 献
- Arai, T., Hasegawa, M., Akiyama, H. et al.: TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun., 351, 602-611 (2006)[PubMed]
- Neumann, M., Sampathu, D. M., Kwong, L. K. et al.: Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science, 314, 130-133 (2006)[PubMed]
- Robberecht, W. & Philips, T.: The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci., 14, 248-264 (2013)[PubMed]
- Sreedharan, J., Blair, I. P., Tripathi, V. B. et al.: TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science, 319, 1668-1672 (2008)[PubMed]
- Kabashi, E., Valdmanis, P. N., Dion, P. et al.: TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet., 40, 572-574 (2008)[PubMed]
- Mackenzie, I. R., Bigio, E. H., Ince, P. G. et al.: Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol., 61, 427-434 (2007)[PubMed]
- Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R. et al.: Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci., 14, 459-468 (2011)[PubMed]
- Tollervey, J. R., Curk, T., Rogelj, B. et al.: Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci., 14, 452-458 (2011)[PubMed]
- Buratti, E. & Baralle, F. E.: Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem., 276, 36337-36343 (2001)[PubMed]
- Ishihara, T., Ariizumi, Y., Shiga, A. et al.: Decreased number of Gemini of coiled bodies and U12 snRNA level in amyotrophic lateral sclerosis. Hum. Mol. Genet., 22, 4136-4147 (2013)[PubMed]
- Kawahara, Y. & Mieda-Sato, A.: TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA, 109, 3347-3352 (2012)[PubMed]
- Wils, H., Kleinberger, G., Janssens, J. et al.: TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA, 107, 3858-3863 (2010)[PubMed]
- Wegorzewska, I., Bell, S., Cairns, N. J. et al.: TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA, 106, 18809-18814 (2009)[PubMed]
- Polymenidou, M. & Cleveland, D. W.: Prion-like spread of protein aggregates in neurodegeneration. J. Exp. Med., 209, 889-893 (2012)[PubMed]
- Nonaka, T., Masuda-Suzukake, M., Arai, T. et al.: Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep., 4, 124-134 (2013)[PubMed]
- Igaz, L. M., Kwong, L. K., Chen-Plotkin, A. et al.: Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J. Biol. Chem., 284, 8516-8524 (2009)[PubMed]
- Guo, W., Chen, Y., Zhou, X. et al.: An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol., 18, 822-830 (2011)[PubMed]
- Watanabe, S., Kaneko, K. & Yamanaka, K.: Accelerated disease onset with stabilized familial amyotrophic lateral sclerosis (ALS)-linked mutant TDP-43 proteins. J. Biol. Chem., 288, 3641-3654 (2013)[PubMed]
- Iguchi, Y., Katsuno, M., Niwa, J. et al.: Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain, 136, 1371-1782 (2013)[PubMed]
- Nishimoto, Y., Nakagawa, S., Hirose, T. et al.: The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain, 6, 31 (2013)[PubMed]
- Vance, C., Rogelj, B., Hortobagyi, T. et al.: Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science, 323, 1208-1211 (2009)[PubMed]
- Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L. et al.: Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science, 323, 1205-1208 (2009)[PubMed]
- Lagier-Tourenne, C., Polymenidou, M., Hutt, K. R. et al.: Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci., 15, 1488-1497 (2012)[PubMed]
- Rogelj, B., Easton, L. E., Bogu, G. K. et al.: Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep., 2, 603 (2012)[PubMed]
- Ishigaki, S., Masuda, A., Fujioka, Y. et al.: Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci. Rep., 2, 529 (2012)[PubMed]
- Dormann, D. & Haass, C.: Fused in sarcoma (FUS): An oncogene goes awry in neurodegeneration. Mol. Cell. Neurosci., 56, 475-486 (2013)[PubMed]
- Belzil, V. V., Langlais, J. S., Daoud, H. et al.: Novel FUS deletion in a patient with juvenile amyotrophic lateral sclerosis. Arch. Neurol., 69, 653-656 (2012)[PubMed]
- Neumann, M., Rademakers, R., Roeber, S. et al.: A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain, 132, 2922-2931 (2009)[PubMed]
- Mitchell, J. C., McGoldrick, P., Vance, C. et al.: Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta. Neuropathol., 125, 273-288 (2013)[PubMed]
- Huang, C., Zhou, H., Tong, J. et al.: FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet., 7, e1002011 (2011)[PubMed]
- Couthouis, J., Hart, M. P., Shorter, J. et al.: A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. USA, 108, 20881-20890 (2011)[PubMed]
- Couthouis, J., Hart, M. P., Erion, R. et al.: Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum. Mol. Genet., 21, 2899-2911 (2012)[PubMed]
- Elden, A. C., Kim, H. J., Hart, M. P. et al.: Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature, 466, 1069-1075 (2010)[PubMed]
- Lee, T., Li, Y. R., Chesi, A. et al.: Evaluating the prevalence of polyglutamine repeat expansions in amyotrophic lateral sclerosis. Neurology, 76, 2062-2065 (2011)[PubMed]
- Kozlov, G., Safaee, N., Rosenauer, A. et al.: Structural basis of binding of P-body-associated proteins GW182 and ataxin-2 by the Mlle domain of poly(A)-binding protein. J. Biol. Chem., 285, 13599-13606 (2010)[PubMed]
- Renton, A. E., Majounie, E., Waite, A. et al.: A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron, 72, 257-268 (2011)[PubMed]
- DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F. et al.: Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron, 72, 245-256 (2011)[PubMed]
- Ishiura, H., Takahashi, Y. Mitsui, J. et al.: C9ORF72 repeat expansion in amyotrophic lateral sclerosis in the Kii peninsula of Japan. Arch. Neurol., 69, 1154-1158 (2012)[PubMed]
- Mori, K., Weng, S. M., Arzberger, T. et al.: The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science, 339, 1335-1338 (2013)[PubMed] [PubMed] [新着論文レビュー]
- Kim, H. J., Kim, N. C., Wang, Y. D. et al.: Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature, 495, 467-473 (2013)[PubMed]
- Watts, G. D., Wymer, J., Kovach, M. J. et al.: Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet., 36, 377-381 (2004)[PubMed]
著者プロフィール
略歴:2004年 東京大学大学院医学系研究科博士課程 修了,同年 米国Wistar Instituteポスドク研究員,2009年 大阪大学大学院医学系研究科 テニュアトラック准教授を経て,2013年 同 独立准教授.
研究テーマ:RNAの機能や修飾の異常を切り口に,筋萎縮性側索硬化症など疾患の病態を解明する“RNA病態学”をメインテーマにあげている.
研究室URL:http://www.med.osaka-u.ac.jp/pub/rna/index.html
© 2013 河原 行郎 Licensed under CC 表示 2.1 日本
