デルタの世界:ある孤児受容体の物語
2014/03/20
柚﨑 通介
(慶應義塾大学医学部 生理学教室)
email:柚﨑通介
領域融合レビュー, 3, e003 (2014) DOI: 10.7875/leading.author.3.e003
Michisuke Yuzaki: Delta receptors: a tale of an orphan receptor.

哺乳類の中枢神経系におけるミリ秒単位の“速い”興奮性の神経伝達は,シナプス前部から放出されるグルタミン酸と,それをシナプス後部にて受容するイオンチャネル型グルタミン酸受容体により担われる.デルタ型受容体は,イオンチャネル型グルタミン酸受容体の一員として,1993年に相同性スクリーニングによりその遺伝子がクローニングされた.しかし,デルタ型受容体はグルタミン酸とは結合せず,イオンチャネルとしての活性も検出できなかった.そのため,長年にわたり“孤児受容体”とされてきた.一方,デルタ型受容体を欠損したマウスの解析から,デルタ型受容体が脳の機能に必須であることは明確であった.したがって,デルタ型受容体がどのように機能するのかについては長年の謎であった.発見後20年をむかえ,ようやく全貌が明らかになってきたデルタ型受容体の研究の歴史を概説する.
イオンチャネル型グルタミン酸受容体には,薬剤への反応性から,AMPA型,NMDA型,カイニン酸型が存在すると生理学的に考えられていた.1989年にAMPA型グルタミン酸受容体GluA1をコードする遺伝子が最初にクローニングされてから2~3年のあいだに,残りのAMPA型受容体(GluA2~GluA4),NMDA型受容体(GluN1,GluN2A~GluN2D),カイニン酸型受容体(GluK1~GluK5)をコードする遺伝子がつぎつぎとクローニングされた.分子生物学の威力とともに,現象から積み上げていく生理学の偉大さを再認識した時代である.デルタ型受容体GluD1およびGluD2をコードする遺伝子はこれらのゴールドラッシュからやや遅れ,1993年に既知のイオンチャネル型グルタミン酸受容体との相同性スクリーニングによりクローニングされた1-3).GluD1およびGluD2はアミノ酸配列からは明らかにイオンチャネル型グルタミン酸受容体に属する.しかし,グルタミン酸をはじめ結合するリガンドはわからず,イオンチャネルとしての活性も不明であったことから,ずっと“孤児受容体”(orphan receptor)とされてきた.GluD2を欠損したマウスは著明な小脳失調症状をきたす4,5).また,GluD1を欠損したマウスには高周波数の聴覚障害がみられる6).このように,孤児受容体でありながら,GluD1およびGluD2は中枢神経系において重要な機能をはたすことが明らかであった.しかし,これらの機能がどのように制御されているのかについては,長年にわたり謎のまま残されていた.
GluD1およびGluD2の研究には日本人研究者が大きく寄与してきた.その結果,ようやくGluD1およびGluD2のリガンドおよびシグナル伝達機構が明らかになってきた.また,近年では,統合失調症あるいは高次機能障害とGluD1あるいはGluD2の遺伝子に生じた変異との関係が指摘されるようになり,GluD1およびGluD2のシグナル伝達機構の理解は,小脳や内耳のみではなく,中枢神経系における一般的なシナプス機能の解明につながるものと期待されている.個人的には,デルタ型受容体の遺伝子がクローニングされた1993年は筆者が大学院博士課程を修了した年であり,筆者自身も,自分の研究をとおしてこの孤児受容体のシグナル伝達の解明の歴史をずっと見守る立場にあった.未解明な点は数多く残されているが,発見後20年あまりの成人式の代わりとして,これまでのデルタ型受容体の研究小史と今後の展望についてまとめる.
プルキンエ細胞は小脳皮質からその外へと唯一の出力を送る神経細胞である.プルキンエ細胞は,樹状突起の遠位部に顆粒細胞からの平行線維,樹状突起の近位部に下オリーブ核からの登上線維の,2種類の興奮性の入力をうける.1個のプルキンエ細胞は10~20万本もの平行線維とシナプスを形成する一方で,1本の登上線維としかシナプスを形成しない.プルキンエ細胞は平行線維により17~150 Hzの頻度で発火する(単純スパイク)が,登上線維からの入力では1~2 Hzしか発火しない(複雑スパイク).しかし,ひとたび登上線維からの入力により発火すると,プルキンエ細胞においてCa2+濃度の大きな上昇をひき起こす.その結果,登上線維と平行線維が同時に入力することがくり返されると平行線維-プルキンエ細胞シナプスにおけるシナプス伝達の効率が長期間にわたり低下する.この現象を長期抑圧(LTD:long-term depression)とよぶ.われわれの脳に蓄えられる記憶にはさまざまな種類が存在するが,技能や運動に関与する記憶の形成には小脳が重要な役割をはたす.記憶の基礎となる過程は,シナプスにおける神経伝達の効率の長期的な変化であると考えられている.小脳における運動学習の基礎となる過程は,平行線維-プルキンエ細胞シナプスにおける長期抑圧であり,登上線維からの入力のタイミングが“教師信号”として平行線維-プルキンエ細胞シナプスにおける長期抑圧の誘導を制御するという学習理論(Marr-Albus-Ito理論)が確立されている7).
GluD2は平行線維-プルキンエ細胞シナプスにおいてプルキンエ細胞のもつ樹状突起の棘突起にほぼ選択的に発現する.GluD2をコードする遺伝子は,ほかのグルタミン酸受容体の遺伝子(平均190 kb)と比べきわめて大きい(約1.4 Mb).また,染色体脆弱部位に位置することから,高率で変異が起こる.このうち,ヌル(無発現)変異を示すマウスをhotfootマウスとよび,これまで少なくとも16種類の独立したhotfoot変異が報告されている8).人工的に作製した遺伝子ノックアウトマウスを含め,これらのGluD2欠損マウスでは平行線維-プルキンエ細胞シナプスの数が正常なマウスの60%くらいまで減少する5,9).その結果,GluD2欠損マウスではプルキンエ細胞の樹状突起にシナプス前部が存在しない“はだかの棘突起”が数多く観察される.X線の照射により顆粒細胞の数および平行線維からの入力を低下させた際にも,このようなはだかの棘突起は一時的に観察される.しかし,この場合には,いずれは残存した平行線維とシナプスを形成する.GluD2欠損マウスの特徴ははだかの棘突起が持続して存在することであり,GluD2は平行線維-プルキンエ細胞シナプスの形成に重要であると予想された.実際に,GluD2欠損マウスのプルキンエ細胞にウイルスベクターによりGluD2を発現させると,平行線維-プルキンエ細胞シナプスは数日のうちに急速に形成した10).また,ヒト胎児の腎臓に由来する細胞であるHEK293細胞にGluD2を発現させ顆粒細胞と共培養すると,HEK293細胞のうえに平行線維-プルキンエ細胞シナプスが異所的に形成された11).一方,マウスが成体になってからGluD2を欠失させると,すでに形成されていた平行線維-プルキンエ細胞シナプスはしだいにはずれ,はだかの棘突起が出現した12).これらの結果から,GluD2はin vitroおよびin vivoにおいて平行線維-プルキンエ細胞シナプスの形成および維持を制御する機能をもつことがわかった.
GluD2欠損マウスでは登上線維-プルキンエ細胞シナプスにも異常がみられる.野生型のマウスにおいてプルキンエ細胞は幼若時には複数本の登上線維とシナプスを形成し,これらは成熟とともに1本の登上線維を残して刈り込まれる.しかし,GluD2欠損マウスのプルキンエ細胞は成熟したのちにも複数本の登上線維とシナプスを形成する“幼若型”を示した5,9).このような幼若型の登上線維による支配は,平行線維とプルキンエ細胞とのあいだのシナプス伝達が低下するマウス,たとえば,代謝型グルタミン酸受容体を欠損したマウスやCa2+チャネルを欠損したマウスにおいても観察される13).したがって,この表現型はGluD2の欠損により平行線維-プルキンエ細胞シナプスが減少することによる,2次的な結果であると考えられる.また,野生型マウスでは登上線維-プルキンエ細胞シナプスはプルキンエ細胞の樹状突起の近位部に限局して形成されるが,GluD2欠損マウスでは近位部にくわえ遠位部の樹状突起にまで登上線維-プルキンエ細胞シナプスが形成される14).この現象も,平行線維-プルキンエ細胞シナプスが減少したため,遠位部の樹状突起にまで登上線維が進入できるようになったと考えることができる.2つの入力をうける神経細胞において,一方の入力線維とのシナプスが減少すると,もうひとつの入力線維がそのシナプスのあった領域をうばう現象は,ほかの脳部位においても観察されるからである.このように,GluD2欠損マウスにおいて観察される登上線維-プルキンエ細胞シナプスの異常は,いずれも平行線維-プルキンエ細胞シナプスの減少による2次的な現象として説明が可能である.
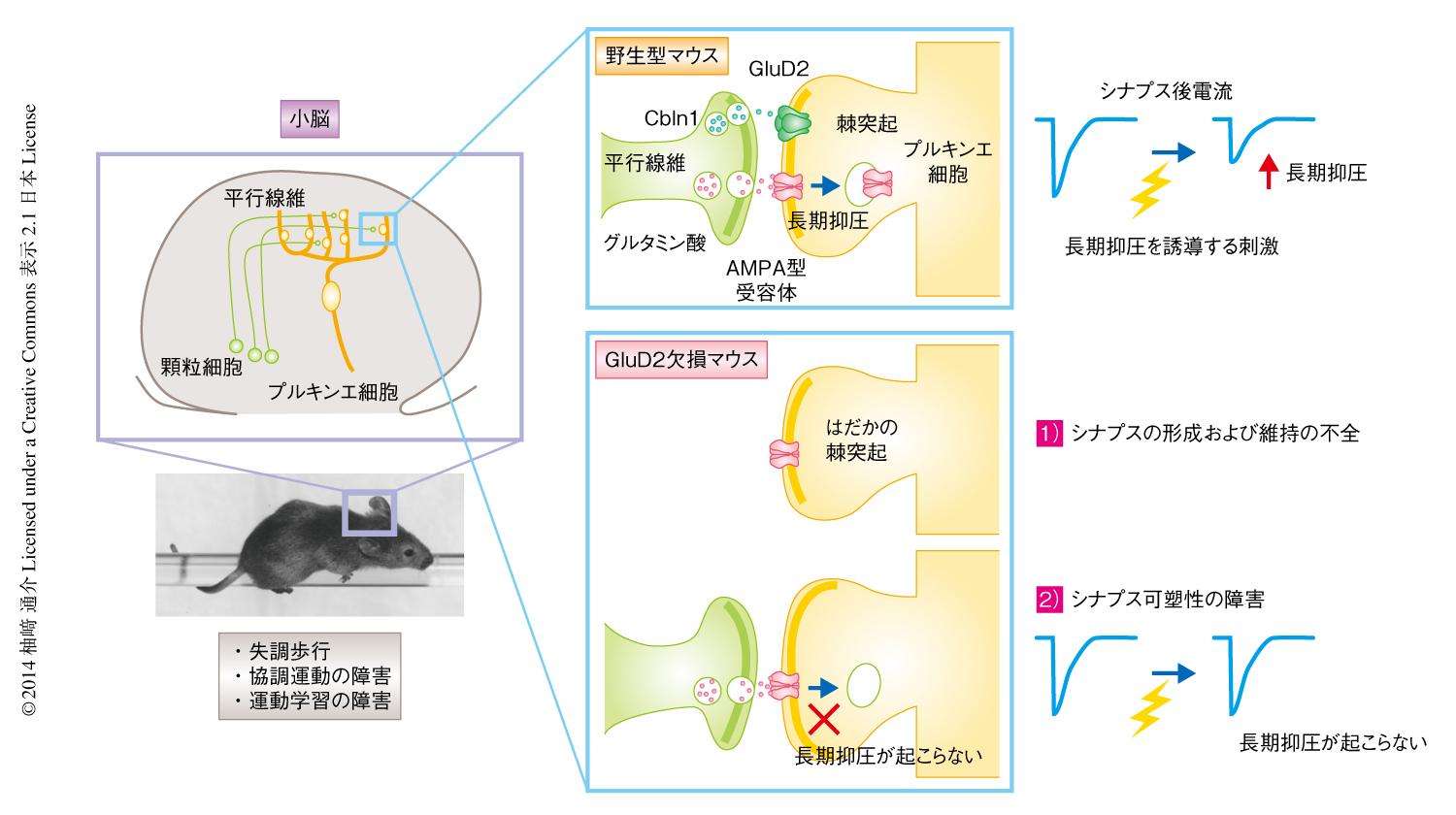
興味深いことに,残存した平行線維-プルキンエ細胞シナプスは形態的には正常であったものの,シナプス可塑性として長期抑圧をひき起こすことができず,機能的には正常ではない4).長期抑圧が起こるためには平行線維からの入力と登上線維からの入力が必要である.しかし,GluD2欠損マウスにおける長期抑圧の誘導の障害は,平行線維あるいは登上線維とプルキンエ細胞とのシナプスの形成異常によるわけではないことも判明した.また,GluD2に対する抗体をプルキンエ細胞に投与すると長期抑圧がひき起こされることもわかった15).つまり,GluD2は,平行線維-プルキンエ細胞シナプスを形成する機能にくわえ,(形態的に完成した)平行線維-プルキンエ細胞シナプスにおいてシナプス可塑性の長期抑圧を制御するという機能をもっていた(図1).では,このようなGluD2の2つの機能は,いったいどのような分子機構により担われるのだろうか?
一般に,リガンドが不明な孤児受容体のシグナル伝達機構を解明することは困難である.逆に,シグナル伝達機構がわからないため,機能スクリーニング法によるリガンドの同定も不可能であった.そこで,筆者らは,さまざまな部位に変異をくわえたGluD2をGluD2欠損マウスのプルキンエ細胞に発現させ,平行線維-プルキンエ細胞シナプスの形成不全と長期抑圧の障害という2つの主要な表現型が回復するかどうかを検討する“レスキュー実験”を行うことにより,これらの機能に関与するGluD2の機能ドメインを絞り込むことからスタートした16).そのため,プルキンエ細胞においてさまざまな変異GluD2を特異的に発現するトランスジェニックマウスを作製しGluD2欠損マウスと交配した.また,ウイルスベクターを用いてGluD2欠損マウスのプルキンエ細胞にさまざまな変異GluD2を発現させることも行った.遠回りのアプローチにみえるかもしれないが,機能ドメインを絞り込むことが孤児受容体GluD2のシグナル伝達機構を探るうえでは近道であろうと考えたのである.
GluD2はほかのイオンチャネル型グルタミン酸受容体と同様の構造をとると考えられる.細胞外から最N末端ドメインとリガンド結合ドメイン,細胞膜の内部にイオンチャネルポアドメイン,そして,細胞内にC末端ドメインをもつ(図2).おもしろいことに,最N末端ドメインを欠失させた変異GluD2をGluD2欠損マウスに発現させると,長期抑圧の障害は回復したものの,平行線維-プルキンエ細胞シナプスの低形成は回復しなかった17).逆に,C末端ドメインの4アミノ酸残基を欠失させた変異GluD2をGluD2欠損マウスに発現させると,平行線維-プルキンエ細胞シナプスの低形成は回復し,登上線維-プルキンエ細胞シナプスの異常も正常化したものの,長期抑圧の障害は回復しなかった10,18,19).一方,リガンド結合ドメイン20) やチャネルポアドメイン21) に変異を導入したGluD2をGluD2欠損マウスに発現させても,平行線維-プルキンエ細胞シナプスの低形成および長期抑圧の障害はともに回復した.これらの結果から,GluD2のもつ2つの主要な機能の発現には,リガンドとの結合やイオンチャネルとしての機能は不要であることが判明した.また,GluD2の最N末端ドメインとC末端ドメインは,それぞれ,平行線維-プルキンエ細胞シナプスの形成および長期抑圧の誘導という2つの機能を別個に制御することが判明したのである22)(図2).逆に,最N末端ドメインとC末端ドメインさえあれば,リガンド結合ドメインやチャネルポアドメインは不要であることも判明した23).それにしても,GluD2はイオンチャネル型グルタミン酸受容体に属するにもかかわらず,イオンチャネルとしてもグルタミン酸の受容体としても機能しないという,かなり変則的なタンパク質であった.どおりで,そのシグナル伝達機構がなかなかわからなかったわけである.

いったい,GluD2は最N末端ドメインを介してどのように平行線維-プルキンエ細胞シナプスの形成を制御しているのだろうか? この答えは,まったく予想外の方向からきた.結論からいうと,顆粒細胞から分泌されるCbln1というこれまでイオンチャネル型グルタミン酸受容体とはまったく無関係と考えられていたタンパク質が,平行線維-プルキンエ細胞シナプスの前部に存在するNeurexinと,シナプス後部に存在するGluD2の最N末端ドメインとに同時に結合することにより,平行線維-プルキンエ細胞シナプスの形成および維持を制御するのである.筆者らが,この発見にいたった経緯を簡単に紹介する.
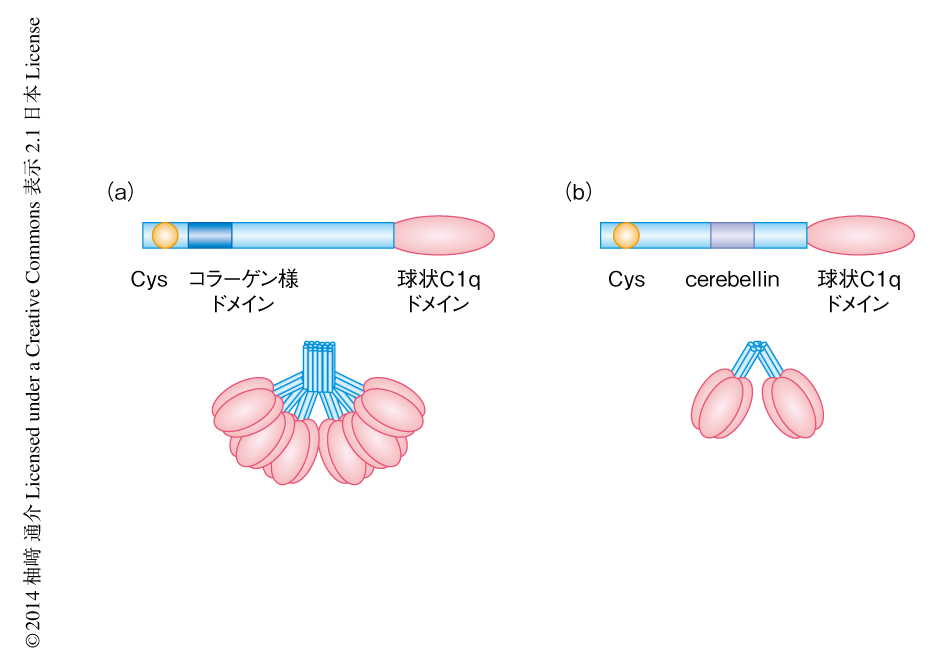
1984年に小脳に特異的なペプチドとしてcerebellinが同定された24).そして1991年にcerebellinをコードするprecerebellin mRNAが同定された25).しかし,precerebellin mRNAがコードするアミノ酸配列にはペプチドの前駆体としてのモチーフは存在せず,全長として自然免疫系の補体C1qと類似したタンパク質がコードされていた.このため,precerebellinはcerebellinの前駆体ではなく,それ自体が機能をもつのではないかと考えられ,“pre”という接頭語は不適当であることからのちにCbln1とよばれることになった(図3).Cbln1は顆粒細胞にほぼ選択的に発現し,そのヌル変異マウスは顆粒細胞の数に軽度の低下(10%)がみられるとともに運動失調を示した.このことから,当初はCbln1は顆粒細胞の生存を維持するタンパク質であると考えられた.しかし,ほかの表現型はみつからず,Cbln1の受容体やシグナル伝達機構は依然として不明であった.
2002年ごろになり,筆者らの研究室にCbln1欠損マウスの解析依頼がもちこまれた.ほどなく,電気生理学的な解析により,Cbln1欠損マウスは平行線維-プルキンエ細胞シナプスの数が低下し,かつ,長期抑圧をひき起こさせないことが判明した26).このことから,Cbln1は平行線維-プルキンエ細胞シナプスにおいて機能することが強く疑われた.そこで,平行線維-プルキンエ細胞シナプスを電子顕微鏡により詳細に解析してもらったところ,シナプスの数の劇的な低下とプルキンエ細胞におけるはだかの棘突起の出現という,GluD2欠損マウスと酷似した表現型をもつことが判明した.また,Cbln1は顆粒細胞から分泌されることもわかった.このように,それぞれの欠損マウスの表現型が酷似することから,受容体が不明なCbln1とリガンドが不明なGluD2は,互いに複合体を形成するのではないかと考えるにいたった.そののち,GluD2の最N末端ドメインにCbln1が特異的に結合することをin vitro結合実験により明らかにすることに成功した.また,免疫組織染色により,平行線維-プルキンエ細胞シナプスのシナプス間隙にはCbln1が局在し,GluD2欠損マウスではシナプス間隙からCbln1が消失することがわかった27).Cbln1欠損マウスにCbln1を投与すると平行線維-プルキンエ細胞シナプスが数日のうちに形成されたが28),Cbln1とGluD2との二重欠損マウスではCbln1の投与によるシナプスの形成はみられなかった27).これらの実験事実は,平行線維から分泌されたCbln1がプルキンエ細胞のGluD2と結合することにより,平行線維-プルキンエ細胞シナプスの形成および維持を制御していことを示した.ほどなく,Cbln1はシナプス後部に存在するGluD2と同時に,シナプス前部に存在するNeurexinとも結合することが判明した29).GluD2のシグナル伝達機構を調べていた筆者らの研究室にCbln1欠損マウスの解析が依頼されたのはまったくの偶然であり,まさかCbln1が長年にわたり探索されていたGluD2のリガンドであるとは,“too good to believe”というべきセレンディピティな発見であった.
この研究を進めるうえで重要な発見のひとつは,GluD2を用いた人工シナプスの形成実験であった11).顆粒細胞と腎臓に由来する非神経細胞であるHEK293細胞とを共培養しても,顆粒細胞の軸索はHEK293細胞にはシナプス前部を形成しない.しかし,GluD2を発現させたHEK293細胞には人工シナプスを形成したのである.筆者らは,Cbln1欠損マウスの顆粒細胞はGluD2を発現するHEK293細胞には人工シナプスを形成しないが,精製したCbln1を培地にくわえることにより人工シナプスの形成を誘導することを見い出した.すなわち,Cbln1は顆粒細胞の内部において作用するのではなく,細胞外に分泌されたCbln1こそがシナプスの形成を誘導することが明確になった.やや遅れて,GluD2を発現したHEK293細胞と顆粒細胞とのあいだに人工シナプスを形成させ,GluD2とともに免疫沈降してくるタンパク質を質量分析法により解析した結果,GluD2,Cbln1,Neurexinからなるタンパク質複合体が形成されることが,筆者らの研究とはまったく独立して報告された30).こうして2010年になり,ついにGluD2は孤児受容体ではなくCbln1の受容体としての位置を確立したのである(図4).

GluD2-Cbln1-Neurexin複合体はこれまでに知られていたシナプス形成タンパク質とは異なる2つの特徴をもっている.ひとつは,シナプスの形態や数そのものに作用する点である.たとえば,Neurexin欠損マウスやNeuroligin欠損マウスではシナプスの数に大きな低下はみられず,シナプスの形成というよりはシナプスの成熟に作用すると考えられている.GluD2-Cbln1-Neurexin複合体もシナプス前部においてシナプス小胞を集積させ,シナプス後部においてGluD2のC末端ドメインに結合するタンパク質を集積させることによりシナプスの成熟に寄与する.これにくわえて,GluD2-Cbln1-Neurexin複合体は平行線維-プルキンエ細胞シナプスの前部から突起を誘導しボタン状の構造を形成させる作用があることがわかった31).NeuroliginもNeurexinと結合するが,このような活性はもたない.この違いは,GluD2と結合したCbln1はNeuroliginよりもNeurexinを高密度に集積させることによるものと考えられている.シナプス形成タンパク質としてのGluD2-Cbln1-Neurexin複合体のもうひとつの大きな特徴は,成熟した個体においてシナプスを形成する活性をもつことである.さきに述べたように,Cbln1欠損マウスの小脳にCbln1を投与すると平行線維-プルキンエ細胞シナプスが数日のうちに形成される.ほかのシナプス形成タンパク質にはin vivoの成熟した脳における活性はないか,あっても非常に弱い.
GluD2はC末端ドメインを介してどのように平行線維-プルキンエ細胞シナプスにおけるシナプス可塑性を制御するのだろうか? イオンチャネル型グルタミン酸受容体のひとつAMPA型受容体が神経活動によってシナプス後部において選択的にエンドサイトーシスされることが,長期抑圧の分子実体であると考えられている.これまでの研究から,平行線維-プルキンエ細胞シナプスにおいて,長期抑圧を誘導する刺激は代謝型グルタミン酸受容体を活性化し,その下流にあるプロテインキナーゼCを活性化し,AMPA型受容体GluA2サブユニットのC末端側に存在する880番目のSerをリン酸化することがわかっていた.GluA2サブユニットはそのC末端側で係留タンパク質GRIPと結合することによりシナプス後部につなぎとめられているが,880番目のSerがリン酸化することによりGRIPからはずれ,別の係留タンパク質PICK1と結合することにより長期抑圧が成立する32)(図5).たとえば,平行線維と登上線維とを同時に刺激する代わりに,小脳の切片にホルボールエステルを投与してプロテインキナーゼCを直接に活性化することによっても,平行線維-プルキンエ細胞シナプスにおいて長期抑圧を誘導することができる33).おもしろいことに,GluD2欠損マウスの小脳の切片にホルボールエステルを投与してもGluA2サブユニットの880番目のSerはリン酸化されず,GRIPがはずれないため長期抑圧は誘導されないことがわかった34).さらに,GluA2サブユニットの880番目のSerの4残基前の876番目にはTyrがあり,GluD2欠損マウスでは定常状態においてこのTyrのリン酸化が亢進していることが判明した.GluA2サブユニットのC末端側の断片を用いたin vitroにおけるリン酸化実験では,876番目のTyrがリン酸化されていると,おそらく電荷あるいは立体的な障害により,880番目のSerのリン酸化が阻害された.また,GluD2欠損マウスの小脳の切片では長期抑圧を誘導する刺激をくわえてもGluA2サブユニットの880番目のSerはリン酸化されず,AMPA型受容体のエンドサイトーシス(つまり,長期抑圧)は起こらなかった.しかし,薬剤処理によりGluA2サブユニットの876番目のTyrを脱リン酸化すると,GluD2欠損マウスにおいても長期抑圧を誘導する刺激によって880番目のSerがリン酸化されるようになり,長期抑圧が回復した.また,876番目のTyrをPheに置換しリン酸化されないようにしたGluA2サブユニットをプルキンエ細胞に発現させたGluD2欠損マウスでは,長期抑圧が正常に誘導された.以上の結果は,AMPA型受容体GluA2サブユニットの876番目のTyrのリン酸化の亢進が,GluD2欠損マウスにおける長期抑圧の障害の原因であることを示している(図5).
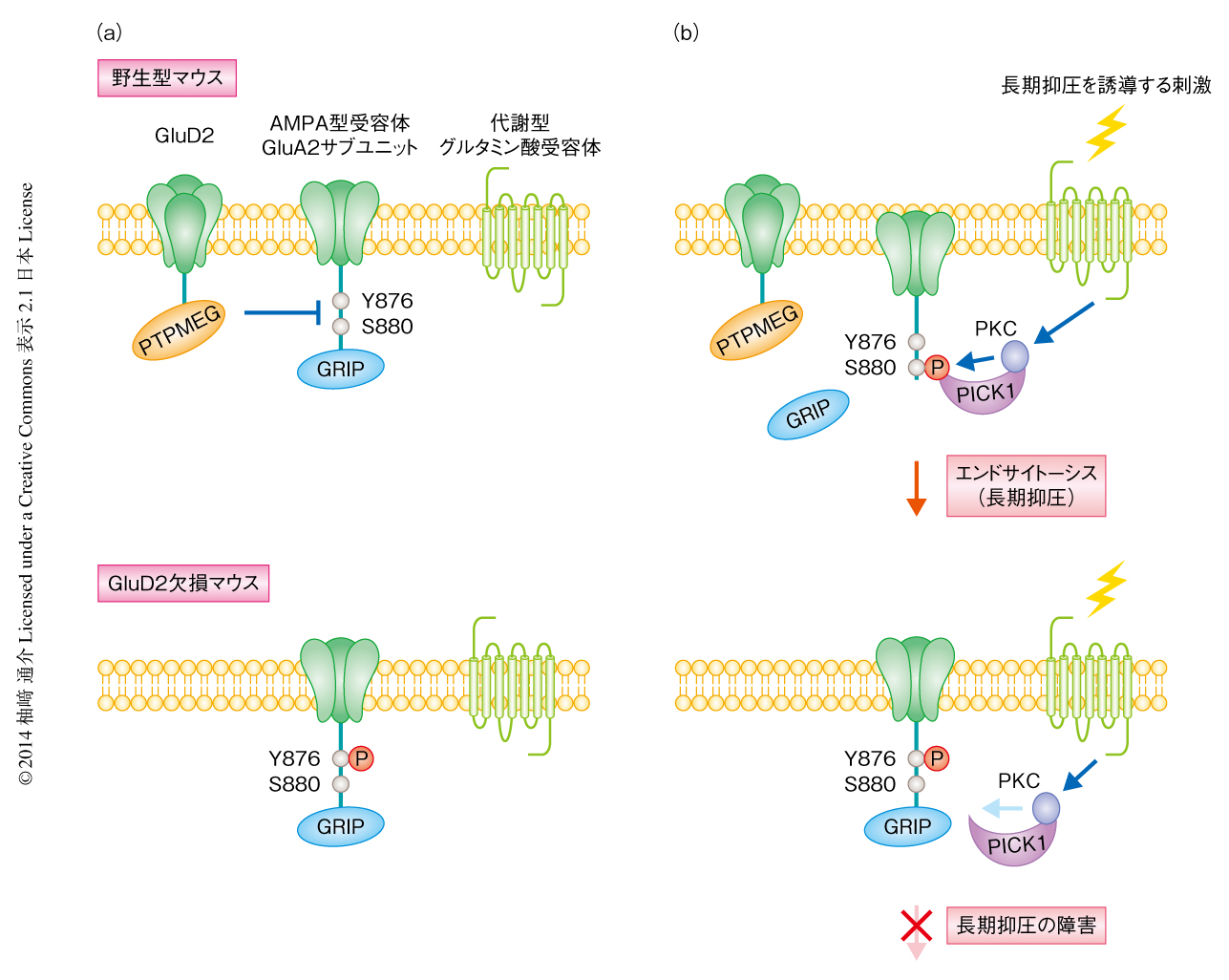
では,GluD2欠損マウスではどうしてAMPA型受容体GluA2サブユニットの876番目のTyrのリン酸化が亢進しているのだろうか? 筆者らは,Tyrのリン酸化に関与し,かつ,プルキンエ細胞に発現してGluD2と結合するタンパク質として,PTPMEGに着目した.PTPMEGはバンド4.1ドメインとPDZドメインをもつ細胞質型チロシンホスファターゼで,2000年に酵母ツーハイブリッド法によりGluD2のC末端ドメインに結合するタンパク質として同定された35).PTPMEG欠損マウスの小脳では長期抑圧が障害されるが36),その分子機構はよくわかっていなかった.筆者らは,GluA2サブユニットの876番目のTyrがPTPMEGの基質として脱リン酸化されることを発見した.実際に,PTPMEG欠損マウスでもこの876番目のTyrのリン酸化が亢進しており,その結果,長期抑圧を誘導する刺激により880番目のSerはリン酸化されなかった.さきに述べたように,C末端ドメインの4アミノ酸残基を欠失させた変異GluD2をGluD2欠損マウスに発現させても長期抑圧の障害は回復しない.しかし,この4アミノ酸残基を欠失させた変異GluD2にPTPMEGのチロシンホスホターゼドメインを直接に融合したキメラGluD2をGluD2欠損マウスに発現させると長期抑圧が回復した.また,ホスファターゼ活性をもたない変異PTPMEGを野生型マウスの小脳プルキンエ細胞に発現させると,内因性PTPMEGの機能を阻害し長期抑圧が障害された.しかし,ホスファターゼ活性をもたないPTPMEGにさらに変異を導入しGluD2と結合できなくした変異PTPMEGは,内因性のPTPMEGの機能を阻害せず長期抑圧も障害しなかった.以上の実験結果から,GluD2のC末端ドメインを介した長期抑圧の制御には,PTPMEGとの結合とそのホスファターゼ活性が必要かつ十分であることが明らかになった34).GluD2のC末端ドメインはPTPMEGと結合することによりAMPA型受容体GluA2サブユニットの876番目のTyrのリン酸化の状態を制御し,“長期抑圧の起こりやすさ”を制御する門番のような役割をはたしていると考えられる37)(図5).
2007年にGluD2のリガンド結合ドメインの結晶構造が解かれ,D-セリンおよびグリシンと結合することにより立体構造が変化することが示された38).しかし,その生理的な意義は不明であった.筆者らは,D-セリンをプルキンエ細胞に投与すると,平行線維-プルキンエ細胞シナプスからAMPA型受容体がエンドサイトーシスされることを見い出した39)(新着論文レビュー でも掲載).GluD2欠損マウスのプルキンエ細胞,あるいは,D-セリンとの結合部位に変異を導入したGluD2を発現するプルキンエ細胞においてはこの現象はみられなかった.また,顆粒細胞の活動により平行線維から放出されたグルタミン酸がBergmannグリアに存在するCa2+透過型のAMPA型受容体に結合すると,BergmannグリアにCa2+が流入しD-セリンが放出されることも見い出された.げっ歯類の小脳においてはD-セリン分解酵素の発現量が生後2~3週より高くなるため,D-セリンの放出は幼若期のみにみられる.生後2~3週までは長期抑圧が誘導されやすく運動学習を獲得しやすいのは,Bergmannグリアから放出されるD-セリンがGluD2のリガンド結合ドメインとの結合を介してAMPA型受容体のエンドサイトーシス(つまり,長期抑圧)をひき起こしやすくするためであることが判明した.このD-セリンとの結合によるAMPA型受容体のエンドサイトーシスの制御にも,GluD2のC末端ドメインは必須であった.このことから,GluD2のリガンド結合ドメインにD-セリンが結合することによりひき起こされるなんらかの立体構造の変化が,細胞内に伝播し,C末端ドメインに結合したPTPMEGを介するAMPA型受容体のエンドサイトーシスを制御する経路を駆動することが考えられる(図4).
このように,GluD2は最N末端ドメインに結合するCbln1により平行線維-プルキンエ細胞シナプスの形成および維持を制御し,リガンド結合ドメインに結合するD-セリンおよびC末端ドメインに結合するPTPMEGによりAMPA型受容体のエンドサイトーシスすなわち長期抑圧を制御することがわかった.重要なことは,これらのいずれの機能もGluD2のイオンチャネルとしての活性を必要としないことである.では,GluD2はイオンチャネルとしてまったく機能しないのであろうか? イオンチャネルとして機能しないとすると,イオンチャネル型グルタミン酸受容体の一員としてチャネルポアドメインのアミノ酸配列が進化的に保存されていることがうまく説明できない.実際に,自然発生したGluD2の遺伝子変異マウスであるlurcherマウスでは,リガンド結合ドメインとチャネルポアドメインとのあいだのリンカー部分の1アミノ酸変異により,GluD2がつねに開口するイオンチャネルとして機能することがわかっている40,41).また,同じイオンチャネル型グルタミン酸受容体であるカイニン酸型受容体あるいはAMPA型受容体のチャネルポアドメインをGluD1あるいはGluD2のチャネルポアドメインと置換したキメラ受容体は,グルタミン酸の投与により開口しイオンチャネル活性を示す42).すなわち,GluD1およびGluD2のチャネルポアドメインは,非生理的な条件ではあるものの,チャネルとして機能することが可能である.
最近になり,代謝型グルタミン酸受容体mGluR1を活性化することにより,生理的な条件においてもGluD2がイオンチャネル活性を示し,時間経過の遅いシナプス後電流をひき起こすことが示された43).また,GluD2はmGluR1やプロテインキナーゼCと複合体を形成しており,GluD2欠損マウスではmGluR1からのシグナル伝達が低下するため遅いシナプス後電流が減弱することも示された44).ただし,これまでにmGluR1が活性した際にみられる遅いシナプス後電流はTRPC3チャネルを通過するイオン流によることが示されており,TRPC3欠損マウスではこの電流はまったく消失する45).このことは,GluD2はイオンチャネルとして機能するのではないことを示唆する.また,遅いシナプス後電流は細胞内におけるチロシンのリン酸化が亢進すると減弱することが知られており,実際に,GluD2欠損マウスにチロシンリン酸化の阻害剤を投与すると,減弱していた遅いシナプス後電流は正常化した.このことから,GluD2欠損マウスにおいて観察される遅いシナプス後電流の減弱は,PTPMEGの活性化の障害によるチロシンリン酸化の亢進によると解釈することができる.このように,GluD2がイオンチャネルとしてはたらくのか,もしはたらくとすれば,どんな場合でありどのような生理的な意義があるのかについては,今後に残された課題である.
近年,GluD2の欠失変異をもつ家系があいついで報告された46-48).これらの患者は,GluD2欠損マウスにおける表現型と同様に,小脳性の運動失調や自発眼振を主徴とする.しかし,2つの予想外の知見がもたらされた.ひとつは,ヒトでは小脳の萎縮がはなはだしい点である.マウスでは小脳の虫部では野生型の12%ほどしか萎縮はみられないが,ヒトでは小脳の虫部で80~96%近くもの容積の減少がみられる.一般に,顆粒細胞の生存のためにはプルキンエ細胞-平行線維シナプスを介した逆行性の栄養因子が必要とされている.ヒトのプルキンエ細胞はマウスと比べ1細胞あたりはるかに多くの平行線維とシナプスを形成している.そのため,GluD2欠損による平行線維-プルキンエ細胞シナプスの減少が顆粒細胞に対してより大きな影響をおよぼすのかもしれない.もうひとつの特徴的な知見は,言語発達の障害である.これは小脳性の構語障害ではなく,言語機能の障害と考えられている.5歳児の症例では2語文しか話せなかったり,12歳児の症例でも3歳児に相当する言語の理解力しかなかったりする47).これらの高次脳機能の障害は小脳の非運動性の機能を反映しているのか,あるいは,小脳以外の脳部位に発現しているGluD2の機能を反映しているのかについては,今後の研究課題となっている.ヒトのGluD2をコードする遺伝子は,マウスと同様に,非常に大きくかつ染色体脆弱部位に存在している.したがって,今後も,GluD2の変異による劣性遺伝性家系や新規の変異による患者が発見されることが予想される.
GluD1はマウスでは内耳の内有毛細胞において強く発現する.幼若期には線条体に強く発現するが,成熟したのちにはおもに海馬において弱い発現が認められるとされてきた2).実際に,GluD1欠損マウスには高周波数の聴覚障害がみられるが,海馬におけるシナプス可塑性は障害されないと報告されていた6).ところが近年,GluD1をコードする遺伝子と精神疾患との関連性があいついで指摘された.たとえば,ゲノムワイド関連解析により双極性障害49,50) あるいは統合失調症51-54),さらに,コピー数多型解析により自閉症との関連性が報告されている.そこで,GluD1欠損マウスの表現型を精査したところ,マウスにおいても不安症状や攻撃性の亢進,うつ症状,社会性の障害がみられることが報告された55).in vitroでの実験において,GluD1の最N末端ドメインにはCbln1やそのファミリータンパク質であるCbln2あるいはCbln4が結合し,シナプス形成能を示した27,56,57).また,アミノ酸配列からはGluD1のC末端ドメインにもPTPMEGが結合することが予想されており,実際に,線条体にはPTPMEGが強く発現している35).これらのことより,GluD1もGluD2と似たシグナル伝達経路を介して前脳において機能し,高次脳機能に関与することが強く予想されている.in vivoにおいてGluD1がどのように作用するのか,これからの研究の進展が待たれる.
遺伝子クローニング,質量分析法,さらに近年では,ゲノム解析により,これまでさまざまな未知のタンパク質が同定されてきた.しかし,同定されたタンパク質の機能の解明は必ずしも簡単ではない.このレビューでは,相同性スクリーニングにより遺伝子クローニングされて以来20年近くの時をへて,ようやくシグナル伝達経路の一端が明らかになってきた元“孤児受容体”デルタ型受容体についての研究小史を記した.あえて独断と我田引水をおそれず振り返ってみると,この研究の進展において,2つの大きなブレイクスルーがあったと思う.ひとつは,レスキュー実験によるGluD2の機能ドメインの絞り込みの成功である.GluD2の最N末端ドメインおよびC末端ドメインがそれぞれ別々の機能に必要かつ十分であることがわかってはじめて,つぎのステップに集中することが可能になった.レスキュー実験はショウジョウバエや線虫においては普通に行われており,また現在では,遺伝子変異マウスの作製や,ウイルスベクターにより特異的に遺伝子を発現させることは,それほどたいへんなことではない.しかし,10年まえの時点では,マウスの表現型を指標としたレスキュー実験の実施にとりかかるには大きな決断を必要とした.このような経験からは,早く結果を求めるばかりでなく,じっくりと腰をすえて研究することの大切さをあらためて実感する.もうひとつの,また,より大きなブレイクスルーは,Cbln1欠損マウスとの出会いである.まったくセレンディピティな発見であったが,GluD2欠損マウスの表現型を知悉していたからこそ,Cbln1がGluD2のリガンドではないかと疑うことができた.結局のところ,未知のタンパク質のシグナル伝達経路の解明は,僥倖を期待しつつ,じっくりこつこつと研究を進めることしかないのかもしれない.
略歴:1993年 自治医科大学大学院博士課程 修了,同年 米国Roche Institute of Molecular Biology研究員,1995年 米国St. Jude Children’s Research Hospital助教授,2002年 同 准教授を経て,2003年より慶應義塾大学医学部 教授.
研究テーマ:記憶など,あらゆる精神現象の素過程の場としてのシナプス可塑性とシナプスの形成および改変の過程を,分子レベルから個体レベルまで一貫して理解したい.
研究室URL:http://www.yuzaki-lab.org/
© 2014 柚﨑 通介 Licensed under CC 表示 2.1 日本
(慶應義塾大学医学部 生理学教室)
email:柚﨑通介
領域融合レビュー, 3, e003 (2014) DOI: 10.7875/leading.author.3.e003
Michisuke Yuzaki: Delta receptors: a tale of an orphan receptor.
要 約
哺乳類の中枢神経系におけるミリ秒単位の“速い”興奮性の神経伝達は,シナプス前部から放出されるグルタミン酸と,それをシナプス後部にて受容するイオンチャネル型グルタミン酸受容体により担われる.デルタ型受容体は,イオンチャネル型グルタミン酸受容体の一員として,1993年に相同性スクリーニングによりその遺伝子がクローニングされた.しかし,デルタ型受容体はグルタミン酸とは結合せず,イオンチャネルとしての活性も検出できなかった.そのため,長年にわたり“孤児受容体”とされてきた.一方,デルタ型受容体を欠損したマウスの解析から,デルタ型受容体が脳の機能に必須であることは明確であった.したがって,デルタ型受容体がどのように機能するのかについては長年の謎であった.発見後20年をむかえ,ようやく全貌が明らかになってきたデルタ型受容体の研究の歴史を概説する.
はじめに
イオンチャネル型グルタミン酸受容体には,薬剤への反応性から,AMPA型,NMDA型,カイニン酸型が存在すると生理学的に考えられていた.1989年にAMPA型グルタミン酸受容体GluA1をコードする遺伝子が最初にクローニングされてから2~3年のあいだに,残りのAMPA型受容体(GluA2~GluA4),NMDA型受容体(GluN1,GluN2A~GluN2D),カイニン酸型受容体(GluK1~GluK5)をコードする遺伝子がつぎつぎとクローニングされた.分子生物学の威力とともに,現象から積み上げていく生理学の偉大さを再認識した時代である.デルタ型受容体GluD1およびGluD2をコードする遺伝子はこれらのゴールドラッシュからやや遅れ,1993年に既知のイオンチャネル型グルタミン酸受容体との相同性スクリーニングによりクローニングされた1-3).GluD1およびGluD2はアミノ酸配列からは明らかにイオンチャネル型グルタミン酸受容体に属する.しかし,グルタミン酸をはじめ結合するリガンドはわからず,イオンチャネルとしての活性も不明であったことから,ずっと“孤児受容体”(orphan receptor)とされてきた.GluD2を欠損したマウスは著明な小脳失調症状をきたす4,5).また,GluD1を欠損したマウスには高周波数の聴覚障害がみられる6).このように,孤児受容体でありながら,GluD1およびGluD2は中枢神経系において重要な機能をはたすことが明らかであった.しかし,これらの機能がどのように制御されているのかについては,長年にわたり謎のまま残されていた.
GluD1およびGluD2の研究には日本人研究者が大きく寄与してきた.その結果,ようやくGluD1およびGluD2のリガンドおよびシグナル伝達機構が明らかになってきた.また,近年では,統合失調症あるいは高次機能障害とGluD1あるいはGluD2の遺伝子に生じた変異との関係が指摘されるようになり,GluD1およびGluD2のシグナル伝達機構の理解は,小脳や内耳のみではなく,中枢神経系における一般的なシナプス機能の解明につながるものと期待されている.個人的には,デルタ型受容体の遺伝子がクローニングされた1993年は筆者が大学院博士課程を修了した年であり,筆者自身も,自分の研究をとおしてこの孤児受容体のシグナル伝達の解明の歴史をずっと見守る立場にあった.未解明な点は数多く残されているが,発見後20年あまりの成人式の代わりとして,これまでのデルタ型受容体の研究小史と今後の展望についてまとめる.
1.ヌル変異マウスが明らかにしたGluD2の古典的な機能
プルキンエ細胞は小脳皮質からその外へと唯一の出力を送る神経細胞である.プルキンエ細胞は,樹状突起の遠位部に顆粒細胞からの平行線維,樹状突起の近位部に下オリーブ核からの登上線維の,2種類の興奮性の入力をうける.1個のプルキンエ細胞は10~20万本もの平行線維とシナプスを形成する一方で,1本の登上線維としかシナプスを形成しない.プルキンエ細胞は平行線維により17~150 Hzの頻度で発火する(単純スパイク)が,登上線維からの入力では1~2 Hzしか発火しない(複雑スパイク).しかし,ひとたび登上線維からの入力により発火すると,プルキンエ細胞においてCa2+濃度の大きな上昇をひき起こす.その結果,登上線維と平行線維が同時に入力することがくり返されると平行線維-プルキンエ細胞シナプスにおけるシナプス伝達の効率が長期間にわたり低下する.この現象を長期抑圧(LTD:long-term depression)とよぶ.われわれの脳に蓄えられる記憶にはさまざまな種類が存在するが,技能や運動に関与する記憶の形成には小脳が重要な役割をはたす.記憶の基礎となる過程は,シナプスにおける神経伝達の効率の長期的な変化であると考えられている.小脳における運動学習の基礎となる過程は,平行線維-プルキンエ細胞シナプスにおける長期抑圧であり,登上線維からの入力のタイミングが“教師信号”として平行線維-プルキンエ細胞シナプスにおける長期抑圧の誘導を制御するという学習理論(Marr-Albus-Ito理論)が確立されている7).
GluD2は平行線維-プルキンエ細胞シナプスにおいてプルキンエ細胞のもつ樹状突起の棘突起にほぼ選択的に発現する.GluD2をコードする遺伝子は,ほかのグルタミン酸受容体の遺伝子(平均190 kb)と比べきわめて大きい(約1.4 Mb).また,染色体脆弱部位に位置することから,高率で変異が起こる.このうち,ヌル(無発現)変異を示すマウスをhotfootマウスとよび,これまで少なくとも16種類の独立したhotfoot変異が報告されている8).人工的に作製した遺伝子ノックアウトマウスを含め,これらのGluD2欠損マウスでは平行線維-プルキンエ細胞シナプスの数が正常なマウスの60%くらいまで減少する5,9).その結果,GluD2欠損マウスではプルキンエ細胞の樹状突起にシナプス前部が存在しない“はだかの棘突起”が数多く観察される.X線の照射により顆粒細胞の数および平行線維からの入力を低下させた際にも,このようなはだかの棘突起は一時的に観察される.しかし,この場合には,いずれは残存した平行線維とシナプスを形成する.GluD2欠損マウスの特徴ははだかの棘突起が持続して存在することであり,GluD2は平行線維-プルキンエ細胞シナプスの形成に重要であると予想された.実際に,GluD2欠損マウスのプルキンエ細胞にウイルスベクターによりGluD2を発現させると,平行線維-プルキンエ細胞シナプスは数日のうちに急速に形成した10).また,ヒト胎児の腎臓に由来する細胞であるHEK293細胞にGluD2を発現させ顆粒細胞と共培養すると,HEK293細胞のうえに平行線維-プルキンエ細胞シナプスが異所的に形成された11).一方,マウスが成体になってからGluD2を欠失させると,すでに形成されていた平行線維-プルキンエ細胞シナプスはしだいにはずれ,はだかの棘突起が出現した12).これらの結果から,GluD2はin vitroおよびin vivoにおいて平行線維-プルキンエ細胞シナプスの形成および維持を制御する機能をもつことがわかった.
GluD2欠損マウスでは登上線維-プルキンエ細胞シナプスにも異常がみられる.野生型のマウスにおいてプルキンエ細胞は幼若時には複数本の登上線維とシナプスを形成し,これらは成熟とともに1本の登上線維を残して刈り込まれる.しかし,GluD2欠損マウスのプルキンエ細胞は成熟したのちにも複数本の登上線維とシナプスを形成する“幼若型”を示した5,9).このような幼若型の登上線維による支配は,平行線維とプルキンエ細胞とのあいだのシナプス伝達が低下するマウス,たとえば,代謝型グルタミン酸受容体を欠損したマウスやCa2+チャネルを欠損したマウスにおいても観察される13).したがって,この表現型はGluD2の欠損により平行線維-プルキンエ細胞シナプスが減少することによる,2次的な結果であると考えられる.また,野生型マウスでは登上線維-プルキンエ細胞シナプスはプルキンエ細胞の樹状突起の近位部に限局して形成されるが,GluD2欠損マウスでは近位部にくわえ遠位部の樹状突起にまで登上線維-プルキンエ細胞シナプスが形成される14).この現象も,平行線維-プルキンエ細胞シナプスが減少したため,遠位部の樹状突起にまで登上線維が進入できるようになったと考えることができる.2つの入力をうける神経細胞において,一方の入力線維とのシナプスが減少すると,もうひとつの入力線維がそのシナプスのあった領域をうばう現象は,ほかの脳部位においても観察されるからである.このように,GluD2欠損マウスにおいて観察される登上線維-プルキンエ細胞シナプスの異常は,いずれも平行線維-プルキンエ細胞シナプスの減少による2次的な現象として説明が可能である.
興味深いことに,残存した平行線維-プルキンエ細胞シナプスは形態的には正常であったものの,シナプス可塑性として長期抑圧をひき起こすことができず,機能的には正常ではない4).長期抑圧が起こるためには平行線維からの入力と登上線維からの入力が必要である.しかし,GluD2欠損マウスにおける長期抑圧の誘導の障害は,平行線維あるいは登上線維とプルキンエ細胞とのシナプスの形成異常によるわけではないことも判明した.また,GluD2に対する抗体をプルキンエ細胞に投与すると長期抑圧がひき起こされることもわかった15).つまり,GluD2は,平行線維-プルキンエ細胞シナプスを形成する機能にくわえ,(形態的に完成した)平行線維-プルキンエ細胞シナプスにおいてシナプス可塑性の長期抑圧を制御するという機能をもっていた(図1).では,このようなGluD2の2つの機能は,いったいどのような分子機構により担われるのだろうか?
2.孤児受容体GluD2のシグナル伝達機構を探る試み
一般に,リガンドが不明な孤児受容体のシグナル伝達機構を解明することは困難である.逆に,シグナル伝達機構がわからないため,機能スクリーニング法によるリガンドの同定も不可能であった.そこで,筆者らは,さまざまな部位に変異をくわえたGluD2をGluD2欠損マウスのプルキンエ細胞に発現させ,平行線維-プルキンエ細胞シナプスの形成不全と長期抑圧の障害という2つの主要な表現型が回復するかどうかを検討する“レスキュー実験”を行うことにより,これらの機能に関与するGluD2の機能ドメインを絞り込むことからスタートした16).そのため,プルキンエ細胞においてさまざまな変異GluD2を特異的に発現するトランスジェニックマウスを作製しGluD2欠損マウスと交配した.また,ウイルスベクターを用いてGluD2欠損マウスのプルキンエ細胞にさまざまな変異GluD2を発現させることも行った.遠回りのアプローチにみえるかもしれないが,機能ドメインを絞り込むことが孤児受容体GluD2のシグナル伝達機構を探るうえでは近道であろうと考えたのである.
GluD2はほかのイオンチャネル型グルタミン酸受容体と同様の構造をとると考えられる.細胞外から最N末端ドメインとリガンド結合ドメイン,細胞膜の内部にイオンチャネルポアドメイン,そして,細胞内にC末端ドメインをもつ(図2).おもしろいことに,最N末端ドメインを欠失させた変異GluD2をGluD2欠損マウスに発現させると,長期抑圧の障害は回復したものの,平行線維-プルキンエ細胞シナプスの低形成は回復しなかった17).逆に,C末端ドメインの4アミノ酸残基を欠失させた変異GluD2をGluD2欠損マウスに発現させると,平行線維-プルキンエ細胞シナプスの低形成は回復し,登上線維-プルキンエ細胞シナプスの異常も正常化したものの,長期抑圧の障害は回復しなかった10,18,19).一方,リガンド結合ドメイン20) やチャネルポアドメイン21) に変異を導入したGluD2をGluD2欠損マウスに発現させても,平行線維-プルキンエ細胞シナプスの低形成および長期抑圧の障害はともに回復した.これらの結果から,GluD2のもつ2つの主要な機能の発現には,リガンドとの結合やイオンチャネルとしての機能は不要であることが判明した.また,GluD2の最N末端ドメインとC末端ドメインは,それぞれ,平行線維-プルキンエ細胞シナプスの形成および長期抑圧の誘導という2つの機能を別個に制御することが判明したのである22)(図2).逆に,最N末端ドメインとC末端ドメインさえあれば,リガンド結合ドメインやチャネルポアドメインは不要であることも判明した23).それにしても,GluD2はイオンチャネル型グルタミン酸受容体に属するにもかかわらず,イオンチャネルとしてもグルタミン酸の受容体としても機能しないという,かなり変則的なタンパク質であった.どおりで,そのシグナル伝達機構がなかなかわからなかったわけである.
3.GluD2の最N末端ドメインを介するシナプスの形成を制御するシグナルの実体
いったい,GluD2は最N末端ドメインを介してどのように平行線維-プルキンエ細胞シナプスの形成を制御しているのだろうか? この答えは,まったく予想外の方向からきた.結論からいうと,顆粒細胞から分泌されるCbln1というこれまでイオンチャネル型グルタミン酸受容体とはまったく無関係と考えられていたタンパク質が,平行線維-プルキンエ細胞シナプスの前部に存在するNeurexinと,シナプス後部に存在するGluD2の最N末端ドメインとに同時に結合することにより,平行線維-プルキンエ細胞シナプスの形成および維持を制御するのである.筆者らが,この発見にいたった経緯を簡単に紹介する.
1984年に小脳に特異的なペプチドとしてcerebellinが同定された24).そして1991年にcerebellinをコードするprecerebellin mRNAが同定された25).しかし,precerebellin mRNAがコードするアミノ酸配列にはペプチドの前駆体としてのモチーフは存在せず,全長として自然免疫系の補体C1qと類似したタンパク質がコードされていた.このため,precerebellinはcerebellinの前駆体ではなく,それ自体が機能をもつのではないかと考えられ,“pre”という接頭語は不適当であることからのちにCbln1とよばれることになった(図3).Cbln1は顆粒細胞にほぼ選択的に発現し,そのヌル変異マウスは顆粒細胞の数に軽度の低下(10%)がみられるとともに運動失調を示した.このことから,当初はCbln1は顆粒細胞の生存を維持するタンパク質であると考えられた.しかし,ほかの表現型はみつからず,Cbln1の受容体やシグナル伝達機構は依然として不明であった.
2002年ごろになり,筆者らの研究室にCbln1欠損マウスの解析依頼がもちこまれた.ほどなく,電気生理学的な解析により,Cbln1欠損マウスは平行線維-プルキンエ細胞シナプスの数が低下し,かつ,長期抑圧をひき起こさせないことが判明した26).このことから,Cbln1は平行線維-プルキンエ細胞シナプスにおいて機能することが強く疑われた.そこで,平行線維-プルキンエ細胞シナプスを電子顕微鏡により詳細に解析してもらったところ,シナプスの数の劇的な低下とプルキンエ細胞におけるはだかの棘突起の出現という,GluD2欠損マウスと酷似した表現型をもつことが判明した.また,Cbln1は顆粒細胞から分泌されることもわかった.このように,それぞれの欠損マウスの表現型が酷似することから,受容体が不明なCbln1とリガンドが不明なGluD2は,互いに複合体を形成するのではないかと考えるにいたった.そののち,GluD2の最N末端ドメインにCbln1が特異的に結合することをin vitro結合実験により明らかにすることに成功した.また,免疫組織染色により,平行線維-プルキンエ細胞シナプスのシナプス間隙にはCbln1が局在し,GluD2欠損マウスではシナプス間隙からCbln1が消失することがわかった27).Cbln1欠損マウスにCbln1を投与すると平行線維-プルキンエ細胞シナプスが数日のうちに形成されたが28),Cbln1とGluD2との二重欠損マウスではCbln1の投与によるシナプスの形成はみられなかった27).これらの実験事実は,平行線維から分泌されたCbln1がプルキンエ細胞のGluD2と結合することにより,平行線維-プルキンエ細胞シナプスの形成および維持を制御していことを示した.ほどなく,Cbln1はシナプス後部に存在するGluD2と同時に,シナプス前部に存在するNeurexinとも結合することが判明した29).GluD2のシグナル伝達機構を調べていた筆者らの研究室にCbln1欠損マウスの解析が依頼されたのはまったくの偶然であり,まさかCbln1が長年にわたり探索されていたGluD2のリガンドであるとは,“too good to believe”というべきセレンディピティな発見であった.
この研究を進めるうえで重要な発見のひとつは,GluD2を用いた人工シナプスの形成実験であった11).顆粒細胞と腎臓に由来する非神経細胞であるHEK293細胞とを共培養しても,顆粒細胞の軸索はHEK293細胞にはシナプス前部を形成しない.しかし,GluD2を発現させたHEK293細胞には人工シナプスを形成したのである.筆者らは,Cbln1欠損マウスの顆粒細胞はGluD2を発現するHEK293細胞には人工シナプスを形成しないが,精製したCbln1を培地にくわえることにより人工シナプスの形成を誘導することを見い出した.すなわち,Cbln1は顆粒細胞の内部において作用するのではなく,細胞外に分泌されたCbln1こそがシナプスの形成を誘導することが明確になった.やや遅れて,GluD2を発現したHEK293細胞と顆粒細胞とのあいだに人工シナプスを形成させ,GluD2とともに免疫沈降してくるタンパク質を質量分析法により解析した結果,GluD2,Cbln1,Neurexinからなるタンパク質複合体が形成されることが,筆者らの研究とはまったく独立して報告された30).こうして2010年になり,ついにGluD2は孤児受容体ではなくCbln1の受容体としての位置を確立したのである(図4).
GluD2-Cbln1-Neurexin複合体はこれまでに知られていたシナプス形成タンパク質とは異なる2つの特徴をもっている.ひとつは,シナプスの形態や数そのものに作用する点である.たとえば,Neurexin欠損マウスやNeuroligin欠損マウスではシナプスの数に大きな低下はみられず,シナプスの形成というよりはシナプスの成熟に作用すると考えられている.GluD2-Cbln1-Neurexin複合体もシナプス前部においてシナプス小胞を集積させ,シナプス後部においてGluD2のC末端ドメインに結合するタンパク質を集積させることによりシナプスの成熟に寄与する.これにくわえて,GluD2-Cbln1-Neurexin複合体は平行線維-プルキンエ細胞シナプスの前部から突起を誘導しボタン状の構造を形成させる作用があることがわかった31).NeuroliginもNeurexinと結合するが,このような活性はもたない.この違いは,GluD2と結合したCbln1はNeuroliginよりもNeurexinを高密度に集積させることによるものと考えられている.シナプス形成タンパク質としてのGluD2-Cbln1-Neurexin複合体のもうひとつの大きな特徴は,成熟した個体においてシナプスを形成する活性をもつことである.さきに述べたように,Cbln1欠損マウスの小脳にCbln1を投与すると平行線維-プルキンエ細胞シナプスが数日のうちに形成される.ほかのシナプス形成タンパク質にはin vivoの成熟した脳における活性はないか,あっても非常に弱い.
4.GluD2のC末端ドメインを介するシナプス可塑性を制御するシグナルの実体
GluD2はC末端ドメインを介してどのように平行線維-プルキンエ細胞シナプスにおけるシナプス可塑性を制御するのだろうか? イオンチャネル型グルタミン酸受容体のひとつAMPA型受容体が神経活動によってシナプス後部において選択的にエンドサイトーシスされることが,長期抑圧の分子実体であると考えられている.これまでの研究から,平行線維-プルキンエ細胞シナプスにおいて,長期抑圧を誘導する刺激は代謝型グルタミン酸受容体を活性化し,その下流にあるプロテインキナーゼCを活性化し,AMPA型受容体GluA2サブユニットのC末端側に存在する880番目のSerをリン酸化することがわかっていた.GluA2サブユニットはそのC末端側で係留タンパク質GRIPと結合することによりシナプス後部につなぎとめられているが,880番目のSerがリン酸化することによりGRIPからはずれ,別の係留タンパク質PICK1と結合することにより長期抑圧が成立する32)(図5).たとえば,平行線維と登上線維とを同時に刺激する代わりに,小脳の切片にホルボールエステルを投与してプロテインキナーゼCを直接に活性化することによっても,平行線維-プルキンエ細胞シナプスにおいて長期抑圧を誘導することができる33).おもしろいことに,GluD2欠損マウスの小脳の切片にホルボールエステルを投与してもGluA2サブユニットの880番目のSerはリン酸化されず,GRIPがはずれないため長期抑圧は誘導されないことがわかった34).さらに,GluA2サブユニットの880番目のSerの4残基前の876番目にはTyrがあり,GluD2欠損マウスでは定常状態においてこのTyrのリン酸化が亢進していることが判明した.GluA2サブユニットのC末端側の断片を用いたin vitroにおけるリン酸化実験では,876番目のTyrがリン酸化されていると,おそらく電荷あるいは立体的な障害により,880番目のSerのリン酸化が阻害された.また,GluD2欠損マウスの小脳の切片では長期抑圧を誘導する刺激をくわえてもGluA2サブユニットの880番目のSerはリン酸化されず,AMPA型受容体のエンドサイトーシス(つまり,長期抑圧)は起こらなかった.しかし,薬剤処理によりGluA2サブユニットの876番目のTyrを脱リン酸化すると,GluD2欠損マウスにおいても長期抑圧を誘導する刺激によって880番目のSerがリン酸化されるようになり,長期抑圧が回復した.また,876番目のTyrをPheに置換しリン酸化されないようにしたGluA2サブユニットをプルキンエ細胞に発現させたGluD2欠損マウスでは,長期抑圧が正常に誘導された.以上の結果は,AMPA型受容体GluA2サブユニットの876番目のTyrのリン酸化の亢進が,GluD2欠損マウスにおける長期抑圧の障害の原因であることを示している(図5).
では,GluD2欠損マウスではどうしてAMPA型受容体GluA2サブユニットの876番目のTyrのリン酸化が亢進しているのだろうか? 筆者らは,Tyrのリン酸化に関与し,かつ,プルキンエ細胞に発現してGluD2と結合するタンパク質として,PTPMEGに着目した.PTPMEGはバンド4.1ドメインとPDZドメインをもつ細胞質型チロシンホスファターゼで,2000年に酵母ツーハイブリッド法によりGluD2のC末端ドメインに結合するタンパク質として同定された35).PTPMEG欠損マウスの小脳では長期抑圧が障害されるが36),その分子機構はよくわかっていなかった.筆者らは,GluA2サブユニットの876番目のTyrがPTPMEGの基質として脱リン酸化されることを発見した.実際に,PTPMEG欠損マウスでもこの876番目のTyrのリン酸化が亢進しており,その結果,長期抑圧を誘導する刺激により880番目のSerはリン酸化されなかった.さきに述べたように,C末端ドメインの4アミノ酸残基を欠失させた変異GluD2をGluD2欠損マウスに発現させても長期抑圧の障害は回復しない.しかし,この4アミノ酸残基を欠失させた変異GluD2にPTPMEGのチロシンホスホターゼドメインを直接に融合したキメラGluD2をGluD2欠損マウスに発現させると長期抑圧が回復した.また,ホスファターゼ活性をもたない変異PTPMEGを野生型マウスの小脳プルキンエ細胞に発現させると,内因性PTPMEGの機能を阻害し長期抑圧が障害された.しかし,ホスファターゼ活性をもたないPTPMEGにさらに変異を導入しGluD2と結合できなくした変異PTPMEGは,内因性のPTPMEGの機能を阻害せず長期抑圧も障害しなかった.以上の実験結果から,GluD2のC末端ドメインを介した長期抑圧の制御には,PTPMEGとの結合とそのホスファターゼ活性が必要かつ十分であることが明らかになった34).GluD2のC末端ドメインはPTPMEGと結合することによりAMPA型受容体GluA2サブユニットの876番目のTyrのリン酸化の状態を制御し,“長期抑圧の起こりやすさ”を制御する門番のような役割をはたしていると考えられる37)(図5).
2007年にGluD2のリガンド結合ドメインの結晶構造が解かれ,D-セリンおよびグリシンと結合することにより立体構造が変化することが示された38).しかし,その生理的な意義は不明であった.筆者らは,D-セリンをプルキンエ細胞に投与すると,平行線維-プルキンエ細胞シナプスからAMPA型受容体がエンドサイトーシスされることを見い出した39)(新着論文レビュー でも掲載).GluD2欠損マウスのプルキンエ細胞,あるいは,D-セリンとの結合部位に変異を導入したGluD2を発現するプルキンエ細胞においてはこの現象はみられなかった.また,顆粒細胞の活動により平行線維から放出されたグルタミン酸がBergmannグリアに存在するCa2+透過型のAMPA型受容体に結合すると,BergmannグリアにCa2+が流入しD-セリンが放出されることも見い出された.げっ歯類の小脳においてはD-セリン分解酵素の発現量が生後2~3週より高くなるため,D-セリンの放出は幼若期のみにみられる.生後2~3週までは長期抑圧が誘導されやすく運動学習を獲得しやすいのは,Bergmannグリアから放出されるD-セリンがGluD2のリガンド結合ドメインとの結合を介してAMPA型受容体のエンドサイトーシス(つまり,長期抑圧)をひき起こしやすくするためであることが判明した.このD-セリンとの結合によるAMPA型受容体のエンドサイトーシスの制御にも,GluD2のC末端ドメインは必須であった.このことから,GluD2のリガンド結合ドメインにD-セリンが結合することによりひき起こされるなんらかの立体構造の変化が,細胞内に伝播し,C末端ドメインに結合したPTPMEGを介するAMPA型受容体のエンドサイトーシスを制御する経路を駆動することが考えられる(図4).
5.基本にもどって:GluD2はイオンチャネルとしてはたらかないのか?
このように,GluD2は最N末端ドメインに結合するCbln1により平行線維-プルキンエ細胞シナプスの形成および維持を制御し,リガンド結合ドメインに結合するD-セリンおよびC末端ドメインに結合するPTPMEGによりAMPA型受容体のエンドサイトーシスすなわち長期抑圧を制御することがわかった.重要なことは,これらのいずれの機能もGluD2のイオンチャネルとしての活性を必要としないことである.では,GluD2はイオンチャネルとしてまったく機能しないのであろうか? イオンチャネルとして機能しないとすると,イオンチャネル型グルタミン酸受容体の一員としてチャネルポアドメインのアミノ酸配列が進化的に保存されていることがうまく説明できない.実際に,自然発生したGluD2の遺伝子変異マウスであるlurcherマウスでは,リガンド結合ドメインとチャネルポアドメインとのあいだのリンカー部分の1アミノ酸変異により,GluD2がつねに開口するイオンチャネルとして機能することがわかっている40,41).また,同じイオンチャネル型グルタミン酸受容体であるカイニン酸型受容体あるいはAMPA型受容体のチャネルポアドメインをGluD1あるいはGluD2のチャネルポアドメインと置換したキメラ受容体は,グルタミン酸の投与により開口しイオンチャネル活性を示す42).すなわち,GluD1およびGluD2のチャネルポアドメインは,非生理的な条件ではあるものの,チャネルとして機能することが可能である.
最近になり,代謝型グルタミン酸受容体mGluR1を活性化することにより,生理的な条件においてもGluD2がイオンチャネル活性を示し,時間経過の遅いシナプス後電流をひき起こすことが示された43).また,GluD2はmGluR1やプロテインキナーゼCと複合体を形成しており,GluD2欠損マウスではmGluR1からのシグナル伝達が低下するため遅いシナプス後電流が減弱することも示された44).ただし,これまでにmGluR1が活性した際にみられる遅いシナプス後電流はTRPC3チャネルを通過するイオン流によることが示されており,TRPC3欠損マウスではこの電流はまったく消失する45).このことは,GluD2はイオンチャネルとして機能するのではないことを示唆する.また,遅いシナプス後電流は細胞内におけるチロシンのリン酸化が亢進すると減弱することが知られており,実際に,GluD2欠損マウスにチロシンリン酸化の阻害剤を投与すると,減弱していた遅いシナプス後電流は正常化した.このことから,GluD2欠損マウスにおいて観察される遅いシナプス後電流の減弱は,PTPMEGの活性化の障害によるチロシンリン酸化の亢進によると解釈することができる.このように,GluD2がイオンチャネルとしてはたらくのか,もしはたらくとすれば,どんな場合でありどのような生理的な意義があるのかについては,今後に残された課題である.
6.デルタ型受容体と高次の脳機能との関係:小脳と内耳をこえて
近年,GluD2の欠失変異をもつ家系があいついで報告された46-48).これらの患者は,GluD2欠損マウスにおける表現型と同様に,小脳性の運動失調や自発眼振を主徴とする.しかし,2つの予想外の知見がもたらされた.ひとつは,ヒトでは小脳の萎縮がはなはだしい点である.マウスでは小脳の虫部では野生型の12%ほどしか萎縮はみられないが,ヒトでは小脳の虫部で80~96%近くもの容積の減少がみられる.一般に,顆粒細胞の生存のためにはプルキンエ細胞-平行線維シナプスを介した逆行性の栄養因子が必要とされている.ヒトのプルキンエ細胞はマウスと比べ1細胞あたりはるかに多くの平行線維とシナプスを形成している.そのため,GluD2欠損による平行線維-プルキンエ細胞シナプスの減少が顆粒細胞に対してより大きな影響をおよぼすのかもしれない.もうひとつの特徴的な知見は,言語発達の障害である.これは小脳性の構語障害ではなく,言語機能の障害と考えられている.5歳児の症例では2語文しか話せなかったり,12歳児の症例でも3歳児に相当する言語の理解力しかなかったりする47).これらの高次脳機能の障害は小脳の非運動性の機能を反映しているのか,あるいは,小脳以外の脳部位に発現しているGluD2の機能を反映しているのかについては,今後の研究課題となっている.ヒトのGluD2をコードする遺伝子は,マウスと同様に,非常に大きくかつ染色体脆弱部位に存在している.したがって,今後も,GluD2の変異による劣性遺伝性家系や新規の変異による患者が発見されることが予想される.
GluD1はマウスでは内耳の内有毛細胞において強く発現する.幼若期には線条体に強く発現するが,成熟したのちにはおもに海馬において弱い発現が認められるとされてきた2).実際に,GluD1欠損マウスには高周波数の聴覚障害がみられるが,海馬におけるシナプス可塑性は障害されないと報告されていた6).ところが近年,GluD1をコードする遺伝子と精神疾患との関連性があいついで指摘された.たとえば,ゲノムワイド関連解析により双極性障害49,50) あるいは統合失調症51-54),さらに,コピー数多型解析により自閉症との関連性が報告されている.そこで,GluD1欠損マウスの表現型を精査したところ,マウスにおいても不安症状や攻撃性の亢進,うつ症状,社会性の障害がみられることが報告された55).in vitroでの実験において,GluD1の最N末端ドメインにはCbln1やそのファミリータンパク質であるCbln2あるいはCbln4が結合し,シナプス形成能を示した27,56,57).また,アミノ酸配列からはGluD1のC末端ドメインにもPTPMEGが結合することが予想されており,実際に,線条体にはPTPMEGが強く発現している35).これらのことより,GluD1もGluD2と似たシグナル伝達経路を介して前脳において機能し,高次脳機能に関与することが強く予想されている.in vivoにおいてGluD1がどのように作用するのか,これからの研究の進展が待たれる.
おわりに
遺伝子クローニング,質量分析法,さらに近年では,ゲノム解析により,これまでさまざまな未知のタンパク質が同定されてきた.しかし,同定されたタンパク質の機能の解明は必ずしも簡単ではない.このレビューでは,相同性スクリーニングにより遺伝子クローニングされて以来20年近くの時をへて,ようやくシグナル伝達経路の一端が明らかになってきた元“孤児受容体”デルタ型受容体についての研究小史を記した.あえて独断と我田引水をおそれず振り返ってみると,この研究の進展において,2つの大きなブレイクスルーがあったと思う.ひとつは,レスキュー実験によるGluD2の機能ドメインの絞り込みの成功である.GluD2の最N末端ドメインおよびC末端ドメインがそれぞれ別々の機能に必要かつ十分であることがわかってはじめて,つぎのステップに集中することが可能になった.レスキュー実験はショウジョウバエや線虫においては普通に行われており,また現在では,遺伝子変異マウスの作製や,ウイルスベクターにより特異的に遺伝子を発現させることは,それほどたいへんなことではない.しかし,10年まえの時点では,マウスの表現型を指標としたレスキュー実験の実施にとりかかるには大きな決断を必要とした.このような経験からは,早く結果を求めるばかりでなく,じっくりと腰をすえて研究することの大切さをあらためて実感する.もうひとつの,また,より大きなブレイクスルーは,Cbln1欠損マウスとの出会いである.まったくセレンディピティな発見であったが,GluD2欠損マウスの表現型を知悉していたからこそ,Cbln1がGluD2のリガンドではないかと疑うことができた.結局のところ,未知のタンパク質のシグナル伝達経路の解明は,僥倖を期待しつつ,じっくりこつこつと研究を進めることしかないのかもしれない.
文 献
- Araki, K., Meguro, H., Kushiya, E. et al.: Selective expression of the glutamate receptor channel δ2 subunit in cerebellar Purkinje cells. Biochem. Biophys. Res. Commun., 197, 1267-1276 (1993)[PubMed]
- Lomeli, H., Sprengel, R., Laurie, D. J. et al.: The rat delta-1 and delta-2 subunits extend the excitatory amino acid receptor family. FEBS Lett., 315, 318-322 (1993)[PubMed]
- Yamazaki, M., Araki, K., Shibata, A. et al.: Molecular cloning of a cDNA encoding a novel member of the mouse glutamate receptor channel family. Biochem. Biophys. Res. Commun., 183, 886-892 (1992)[PubMed]
- Kashiwabuchi, N., Ikeda, K., Araki, K. et al.: Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluRδ2 mutant mice. Cell, 81, 245-252 (1995)[PubMed]
- Lalouette, A., Lohof, A., Sotelo, C. et al.: Neurobiological effects of a null mutation depend on genetic context: comparison between two hotfoot alleles of the delta-2 ionotropic glutamate receptor. Neuroscience, 105, 443-455 (2001)[PubMed]
- Gao, J., Maison, S. F., Wu, X. et al.: Orphan glutamate receptor delta1 subunit required for high-frequency hearing. Mol. Cell. Biol., 27, 4500-4512 (2007)[PubMed]
- Ito, M.: Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol. Rev., 81, 1143-1195 (2001)[PubMed]
- Wang, Y., Matsuda, S., Drews, V. et al.: A hot spot for hotfoot mutations in the gene encoding the δ2 glutamate receptor. Eur. J. Neurosci., 17, 1581-1590 (2003)[PubMed]
- Kurihara, H., Hashimoto, K., Kano, M. et al.: Impaired parallel fiber→Purkinje cell synapse stabilization during cerebellar development of mutant mice lacking the glutamate receptor δ2 subunit. J. Neurosci., 17, 9613-9623 (1997)[PubMed]
- Kohda, K., Kakegawa, W., Matsuda, S. et al.: The extreme C-terminus of GluRδ2 is essential for induction of long-term depression in cerebellar slices. Eur. J. Neurosci., 25, 1357-1362 (2007)[PubMed]
- Kuroyanagi, T., Yokoyama, M. & Hirano, T.: Postsynaptic glutamate receptor δ family contributes to presynaptic terminal differentiation and establishment of synaptic transmission. Proc. Natl. Acad. Sci. USA, 106, 4912-4916 (2009)[PubMed]
- Takeuchi, T., Miyazaki, T., Watanabe, M. et al.: Control of synaptic connection by glutamate receptor δ2 in the adult cerebellum. J. Neurosci., 25, 2146-2156 (2005)[PubMed]
- Hashimoto, K., Ichikawa, R., Takechi, H. et al.: Roles of glutamate receptor δ2 subunit (GluRδ2) and metabotropic glutamate receptor subtype 1 (mGluR1) in climbing fiber synapse elimination during postnatal cerebellar development. J. Neurosci., 21, 9701-9712 (2001)[PubMed]
- Ichikawa, R., Miyazaki, T., Kano, M. et al.: Distal extension of climbing fiber territory and multiple innervation caused by aberrant wiring to adjacent spiny branchlets in cerebellar Purkinje cells lacking glutamate receptor δ2. J. Neurosci., 22, 8487-8503 (2002)[PubMed]
- Hirai, H., Launey, T., Mikawa, S. et al.: New role of δ2-glutamate receptors in AMPA receptor trafficking and cerebellar function. Nat. Neurosci., 6, 869-876 (2003)[PubMed]
- Yuzaki, M.: Transgenic rescue for characterizing orphan receptors: a review of δ2 glutamate receptor. Transgenic Res., 14, 117-121 (2005)[PubMed]
- Kakegawa, W., Miyazaki, T., Kohda, K. et al.: The N-terminal domain of GluD2 (GluRδ2) recruits presynaptic terminals and regulates synaptogenesis in the cerebellum in vivo. J. Neurosci., 29, 5738-5748 (2009)[PubMed]
- Uemura, T., Kakizawa, S., Yamasaki, M. et al.: Regulation of long-term depression and climbing fiber territory by glutamate receptor δ2 at parallel fiber synapses through its C-terminal domain in cerebellar Purkinje cells. J. Neurosci., 27, 12096-12108 (2007)[PubMed]
- Kakegawa, W., Miyazaki, T., Emi, K. et al.: Differential regulation of synaptic plasticity and cerebellar motor learning by the C-terminal PDZ-binding motif of GluRδ2. J. Neurosci., 28, 1460-1468 (2008)[PubMed]
- Hirai, H., Miyazaki, T., Kakegawa, W. et al.: Rescue of abnormal phenotypes of the δ2 glutamate receptor-null mice by mutant δ2 transgenes. EMBO Rep., 6, 90-95 (2005)[PubMed]
- Kakegawa, W., Kohda, K. & Yuzaki, M.: The δ2 'ionotropic' glutamate receptor functions as a non-ionotropic receptor to control cerebellar synaptic plasticity. J. Physiol., 584, 89-96 (2007)[PubMed]
- Yuzaki, M.: New (but old) molecules regulating synapse integrity and plasticity: Cbln1 and the delta2 glutamate receptor. Neuroscience, 162, 633-643 (2009)[PubMed]
- Torashima, T., Iizuka, A., Horiuchi, H. et al.: Rescue of abnormal phenotypes in δ2 glutamate receptor-deficient mice by the extracellular N-terminal and intracellular C-terminal domains of the δ2 glutamate receptor. Eur. J. Neurosci., 30, 355-365 (2009)[PubMed]
- Slemmon, J. R., Blacher, R., Danho, W. et al.: Isolation and sequencing of two cerebellum-specific peptides. Proc. Natl. Acad. Sci. USA, 81, 6866-6870 (1984)[PubMed]
- Urade, Y., Oberdick, J., Molinar-Rode, R. et al.: Precerebellin is a cerebellum-specific protein with similarity to the globular domain of complement C1q B chain. Proc. Natl. Acad. Sci. USA, 88, 1069-1073 (1991)[PubMed]
- Hirai, H., Pang, Z., Bao, D. et al.: Cbln1 is essential for synaptic integrity and plasticity in the cerebellum. Nat. Neurosci., 8, 1534-1541 (2005)[PubMed]
- Matsuda, K., Miura, E., Miyazaki, T. et al.: Cbln1 is a ligand for an orphan glutamate receptor δ2, a bidirectional synapse organizer. Science, 328, 363-368 (2010)[PubMed]
- Ito-Ishida, A., Miura, E., Emi, K. et al.: Cbln1 regulates rapid formation and maintenance of excitatory synapses in mature cerebellar Purkinje cells in vitro and in vivo. J. Neurosci., 28, 5920-5930 (2008)[PubMed]
- Matsuda, K. & Yuzaki, M.: Cbln family proteins promote synapse formation by regulating distinct neurexin signaling pathways in various brain regions. Eur. J. Neurosci., 33, 1447-1461 (2011)[PubMed]
- Uemura, T., Lee, S. J., Yasumura, M. et al.: Trans-synaptic interaction of GluRδ2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell, 141, 1068-1079 (2010)[PubMed]
- Ito-Ishida, A., Miyazaki, T., Miura, E. et al.: Presynaptically released Cbln1 induces dynamic axonal structural changes by interacting with GluD2 during cerebellar synapse formation. Neuron, 76, 549-564 (2012)[PubMed]
- Matsuda, S., Launey, T., Mikawa, S. et al.: Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J., 19, 2765-2774 (2000)[PubMed]
- Kondo, T., Kakegawa, W. & Yuzaki, M.: Induction of long-term depression and phosphorylation of the delta2 glutamate receptor by protein kinase C in cerebellar slices. Eur. J. Neurosci., 22, 1817-1820 (2005)[PubMed]
- Kohda, K., Kakegawa, W., Matsuda, S. et al.: The δ2 glutamate receptor gates long-term depression by coordinating interactions between two AMPA receptor phosphorylation sites. Proc. Natl. Acad. Sci. USA, 110, E948-E957 (2013)[PubMed]
- Hironaka, K., Umemori, H., Tezuka, T. et al.: The protein-tyrosine phosphatase PTPMEG interacts with glutamate receptor δ2 and ε subunits. J. Biol. Chem., 275, 16167-16173 (2000)[PubMed]
- Kina, S., Tezuka, T., Kusakawa, S. et al.: Involvement of protein-tyrosine phosphatase PTPMEG in motor learning and cerebellar long-term depression. Eur. J. Neurosci., 26, 2269-2278 (2007)[PubMed]
- Yuzaki, M.: Cerebellar LTD vs. motor learning-lessons learned from studying GluD2. Neural Netw., 47, 36-41 (2013)[PubMed]
- Naur, P., Hansen, K. B., Kristensen, A. S. et al.: Ionotropic glutamate-like receptor δ2 binds D-serine and glycine. Proc. Natl. Acad. Sci. USA, 104, 14116-14121 (2007)[PubMed]
- Kakegawa, W., Miyoshi, Y., Hamase, K. et al. .: D-serine regulates cerebellar LTD and motor coordination through the δ2 glutamate receptor. Nat. Neurosci., 14, 603-611 (2011)[PubMed] [新着論文レビュー]
- Zuo, J., De Jager, P. L., Takahashi, K. A. et al.: Neurodegeneration in Lurcher mice caused by mutation in δ2 glutamate receptor gene. Nature, 388, 769-773 (1997)[PubMed]
- Kohda, K., Wang, Y. & Yuzaki, M.: Mutation of a glutamate receptor motif reveals its role in gating and δ2 receptor channel properties. Nat. Neurosci., 3, 315-322 (2000)[PubMed]
- Orth, A., Tapken, D. & Hollmann, M.: The delta subfamily of glutamate receptors: characterization of receptor chimeras and mutants. Eur. J. Neurosci., 37, 1620-1630 (2013)[PubMed]
- Ady, V., Perroy, J., Tricoire, L. et al.: Type 1 metabotropic glutamate receptors (mGlu1) trigger the gating of GluD2 delta glutamate receptors. EMBO Rep., 15, 103-109 (2014)[PubMed]
- Kato, A. S., Knierman, M. D., Siuda, E. R. et al.: Glutamate receptor δ2 associates with metabotropic glutamate receptor 1 (mGluR1), protein kinase Cγ, and canonical transient receptor potential 3 and regulates mGluR1-mediated synaptic transmission in cerebellar Purkinje neurons. J. Neurosci., 32, 15296-15308 (2012)[PubMed]
- Hartmann, J., Dragicevic, E., Adelsberger, H. et al.: TRPC3 channels are required for synaptic transmission and motor coordination. Neuron, 59, 392-398 (2008)[PubMed]
- Utine, G. E., Haliloglu, G., Salanci, B. et al.: A homozygous deletion in GRID2 causes a human phenotype with cerebellar ataxia and atrophy. J. Child Neurol., 28, 926-932 (2013)[PubMed]
- Hills, L. B., Masri, A., Konno, K. et al.: Deletions in GRID2 lead to a recessive syndrome of cerebellar ataxia and tonic upgaze in humans. Neurology, 81, 1378-1386 (2013)[PubMed]
- Maier, A., Klopocki, E., Horn, D. et al.: De novo partial deletion in GRID2 presenting with complicated spastic paraplegia. Muscle Nerve, 49, 289-292 (2014)[PubMed]
- Fallin, M. D., Lasseter, V. K., Avramopoulos, D. et al.: Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am. J. Hum. Genet., 77, 918-936 (2005)[PubMed]
- Venken, T., Alaerts, M., Souery, D. et al.: Chromosome 10q harbors a susceptibility locus for bipolar disorder in Ashkenazi Jewish families. Mol. Psychiatry, 13, 442-450 (2008)[PubMed]
- Guo, S. Z., Huang, K., Shi, Y. Y. et al.: A case-control association study between the GRID1 gene and schizophrenia in the Chinese Northern Han population. Schizophr. Res., 93, 385-390 (2007)[PubMed]
- Treutlein, J., Muhleisen, T. W., Frank, J. et al.: Dissection of phenotype reveals possible association between schizophrenia and Glutamate Receptor Delta 1 (GRID1) gene promoter. Schizophr. Res., 111, 123-130 (2009)[PubMed]
- Glessner, J. T., Wang, K., Cai, G. et al.: Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature, 459, 569-573 (2009)[PubMed]
- Greenwood, T. A., Lazzeroni, L. C., Murray, S. S. et al.: Analysis of 94 candidate genes and 12 endophenotypes for schizophrenia from the Consortium on the Genetics of Schizophrenia. Am. J. Psychiatry, 168, 930-946 (2011)[PubMed]
- Yadav, R., Gupta, S. C., Hillman, B. G. et al.: Deletion of glutamate delta-1 receptor in mouse leads to aberrant emotional and social behaviors. PLoS One, 7, e32969 (2012)[PubMed]
- Ryu, K., Yokoyama, M., Yamashita, M. et al.: Induction of excitatory and inhibitory presynaptic differentiation by GluD1. Biochem. Biophys. Res. Commun., 417, 157-161 (2012)[PubMed]
- Yasumura, M., Yoshida, T., Lee, S. J. et al.: Glutamate receptor δ1 induces preferentially inhibitory presynaptic differentiation of cortical neurons by interacting with neurexins through cerebellin precursor protein subtypes. J. Neurochem., 121, 705-716 (2012)[PubMed]
著者プロフィール
略歴:1993年 自治医科大学大学院博士課程 修了,同年 米国Roche Institute of Molecular Biology研究員,1995年 米国St. Jude Children’s Research Hospital助教授,2002年 同 准教授を経て,2003年より慶應義塾大学医学部 教授.
研究テーマ:記憶など,あらゆる精神現象の素過程の場としてのシナプス可塑性とシナプスの形成および改変の過程を,分子レベルから個体レベルまで一貫して理解したい.
研究室URL:http://www.yuzaki-lab.org/
© 2014 柚﨑 通介 Licensed under CC 表示 2.1 日本
