オートファジーと疾患
2014/06/12
蔭山 俊・小松雅明
(新潟大学大学院医歯学総合研究科 分子生物学)
email:小松雅明
領域融合レビュー, 3, e006 (2014) DOI: 10.7875/leading.author.3.e006
Shun Kageyama & Masaaki Komatsu: Autophagy and disease states.

オートファジー関連遺伝子が同定されて以来,さまざまなモデル動物においてオートファジー関連遺伝子の改変動物が作出され,オートファジーの生理機能,とくに,アミノ酸プールの維持や細胞の新陳代謝としての機能が明確になった.また,さまざまな臓器に特異的なオートファジー関連遺伝子の改変マウスが,変性タンパク質そして異常なオルガネラの蓄積をともなう神経変性疾患,肝障害,がんなど重篤な疾患を呈することが明らかになり,オートファジーによる細胞保護の作用が決定的になった.さらに,細胞に侵入した細菌や異常なオルガネラ,とくに変性ミトコンドリアのオートファジーによる選択的な除去機構が明らかにされ,その病態生理的な役割は複雑さを増している.そしてごく最近,ヒトにおいてもオートファジー関連遺伝子の変異が神経変性疾患や炎症性疾患をひき起こすことが発見され,オートファジーの異常がヒトの疾患にも直接に関与することが判明した.このレビューではオートファジーを概説するとともに,オートファジー関連遺伝子の変異により起こる遺伝性疾患,オートファジーの異常が複雑に関与するがんをとりあげる.
オートファジーはリソソームにおいて細胞質の成分を分解する機構である.オートファジーにはミクロオートファジー,シャペロン介在性オートファジー,マクロオートファジーの3つの経路がある.これらのなかで,マクロオートファジーの解析がもっとも進んでおり,たんに“オートファジー”と表記した場合にはマクロオートファジーをさすことが多く,このレビューにおいてもこれにしたがう.オートファジーは細胞質に出現した隔離膜が細胞質の成分を取り囲みオートファゴソームが形成される過程と,オートファゴソームがリソソームと融合し細胞質の成分を分解する過程からなる.小胞体,ミトコンドリア,ゴルジ体,細胞膜,さらには,リサイクリングエンドソームに由来する多彩な膜成分がオートファゴソーム膜の形成に寄与しており,その形成から内容物の分解までは,オートファジー関連遺伝子(autophagy-related gene:ATG)にコードされるAtgタンパク質により厳密に制御されている.
オートファゴソームの形成に特化したコアAtgタンパク質は,以下の6つの機能単位に分類される.1)ULK1キナーゼ複合体(ULK1,ULK2,Atg13,FIP200,Atg101),2)クラスIIIホスファチジルイノシトール3-キナーゼ(PI3キナーゼ)複合体(Vps34,Vps15,Beclin 1,Atg14(L)/Barkor),3)ホスファチジルイノシトール-1,4,5-トリスリン酸(PI3P)結合タンパク質(Atg2,WIPI1~WIPI4),4)膜タンパク質(Atg9L1),5)Atg12共有結合系(Atg12,Atg7,Atg10,Atg5,Atg16L1,Atg16L2),6)LC3共有結合系(LC3A~LC3C,GABARAP,GABARAPL1,GABARAPL2,Atg4A~Atg4D,Atg7,Atg3).これらコアAtgタンパク質の機能単位が連続的かつ協調的に作動することによりオートファゴソームは形成される1)(図1).
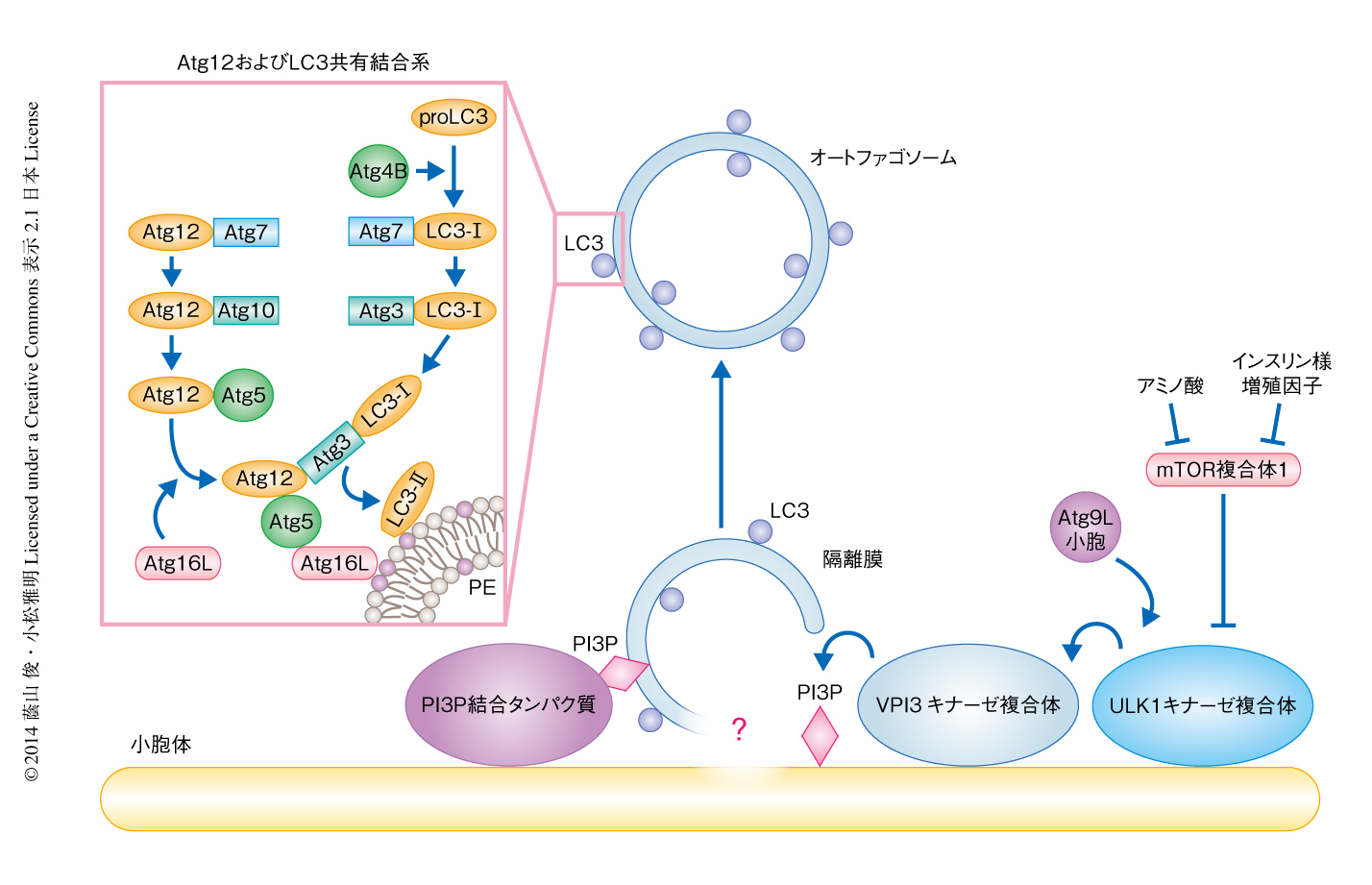
なお,出芽酵母におけるオートファジーの研究については,荒木保弘・大隅良典, 領域融合レビュー, 1, e005, 2012 も参照されたい.
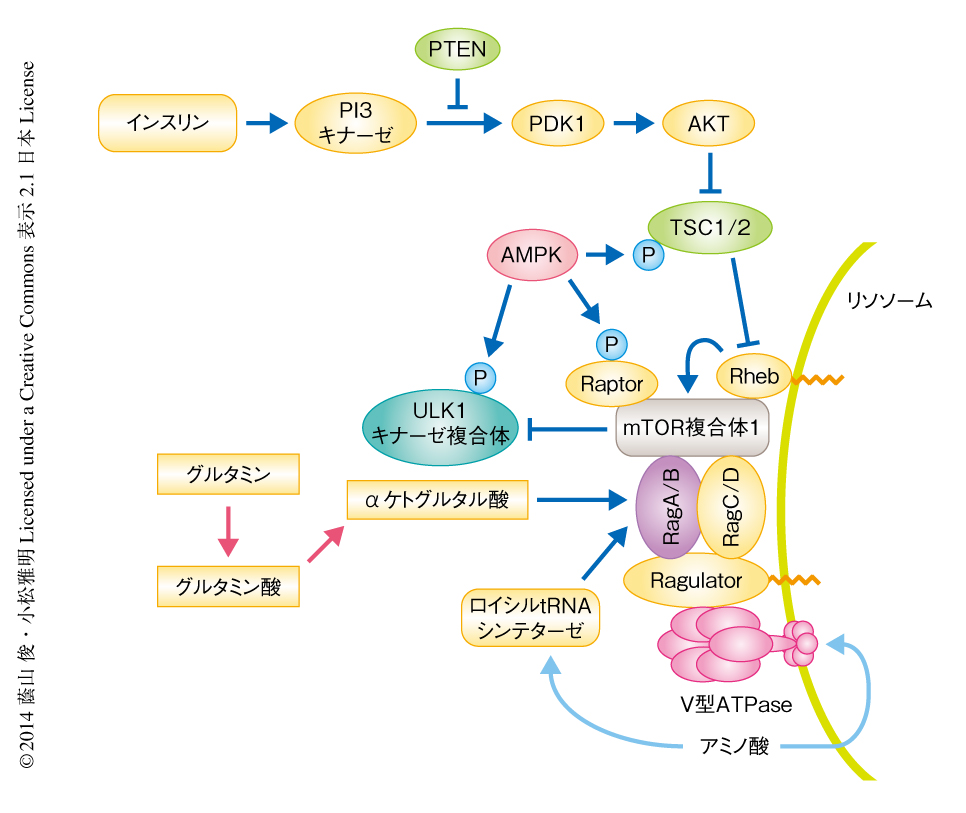
オートファジーはおもにインスリンシグナルおよびアミノ酸シグナルに応じたmTOR複合体1の活性化により抑制される2-4).インスリンシグナルはクラスI PI3キナーゼ-AKT経路を活性化し,TSC2をリン酸化することによりその活性を抑制する.TSC2はGTPase活性化タンパク質(GTPase activating protein:GAP)として低分子量Gタンパク質であるRhebの活性を負に制御する5-8).したがって,AKTによるTSC2の抑制はmTOR複合体1の活性化を促進する(図2).一方,アミノ酸シグナル(とくに,ロイシンおよびグルタミン)により低分子量Gタンパク質であるRagがGDP結合型からGTP結合型へと変換されると,RagはmTOR複合体1の構成タンパク質であるRaptorと結合する.さらに,Ragはリソソーム膜に局在するRagulatorとよばれる複合体と相互作用することによりmTOR複合体1をリソソーム膜につなぎとめる.最終的に,リソソーム膜に存在するRhebとmTOR複合体1とが会合することによりmTOR複合体1の活性化が促進される9-11).ロイシルtRNAシンテターゼはRagのGTPase活性化タンパク質として機能し,ロイシンに誘導されるmTOR複合体1の活性化に寄与する.グルタミンはグルタミナーゼによりグルタミン酸に変換され,さらに,グルタミン酸デヒドロゲナーゼによる脱アミノ化によりαケトグルタル酸とアンモニアになる.αケトグルタル酸はRagのGTP結合型への変換を促進しmTOR複合体1を活性化する12-14).このとき,ロイシンはグルタミン酸デヒドロゲナーゼをアロステリックに活性化している.他方,リソソームに存在するアミノ酸はリソソーム膜に局在するV型ATPaseが感知し,V型ATPaseとRagulatorとの会合あるいは解離を制御することによりmTOR複合体1の活性を制御する15,16).活性化したmTOR複合体1はULK1キナーゼ複合体と相互作用し,ULK1およびAtg13がリン酸化されオートファジーは抑制される17)(図2).すなわち,インスリンあるいはアミノ酸が欠乏するとオートファジーは容易に惹起される(図2).骨格筋ではインスリンにより,一方,肝臓ではアミノ酸によりオートファジーが抑制されており,生体においてオートファジーはmTOR複合体1により組織ごとに異なった分子機構により制御されている18).グルコースが欠乏するとATPが減少してAMP/ATP比が上昇し,その結果,AMPKが活性化する.AMPKはRaptorのリン酸化あるいはTSC2のリン酸化を介してmTOR複合体1を不活性化させオートファジーを誘導する19,20).また,AMPKはULK1およびBeclin 1を直接にリン酸化し活性化する21,22).したがって,AMPKはmTOR複合体1に依存的にも非依存的にオートファジーを活性化しうる(図2).
オートファジーのもっとも基本的な生理機能は,飢餓への適応である(図3a).すでに述べたとおり,オートファジーは低アミノ酸状態あるいは低インスリン状態において一過的に誘導され細胞質の成分を分解する.糖や脂質と異なりアミノ酸プールはわずかであり(ヒトの成人の場合,タンパク質生合成に必要なアミノ酸量は1日あたり300~400 gであるのに対し,アミノ酸プールは約100 gしかない),飢餓に応答したオートファジーによるアミノ酸の供給が重要であることは容易に想像できる.たとえば,マウスの新生仔は胎盤からの栄養の供給が絶たれる出生の直後に一過的にオートファジーを誘導しアミノ酸プールを維持する.全身においてAtg5遺伝子を欠損させたマウスは出生の直後の飢餓の際に組織および血中において重篤な低アミノ酸症状におちいり,それが一因となり出生から24時間以内に死亡する23).では,オートファジーにより生じたアミノ酸はどうなるのだろうか? 出芽酵母のオートファジー不能変異体は飢餓の際にミトコンドリア呼吸鎖タンパク質や抗酸化タンパク質の生合成の不全をひき起こすことから,第1に,飢餓に応答した特定のタンパク質の生合成のために利用されると考えられる.また,アミノ酸はアミノ基転移反応やアミノ酸代謝によりピルビン酸,TCA回路中間体,アセチルCoAとなる.アミノ酸はこの補充反応経路により糖新生,脂肪酸生合成,エネルギー産生に寄与する.実際に,肝臓に特異的なAtg7ノックアウトマウスの解析から,オートファジーにより生じたアミノ酸は糖新生に利用されることが明らかになっている24).がん細胞ではオートファジーに依存したアミノ酸およびTCA回路中間体の供給がその生存に重要らしい25).また,Atg5ノックアウトマウスにおいてはAMPKが活性化されており,糖新生が阻害されていることが示唆される23).受精の直後にもオートファジーは活性化され,母方に由来するタンパク質を分解し,胚の初期発生に必要なタンパク質の生合成のためのアミノ酸を供給する26).このようなオートファジーは,細胞質の成分を非選択的かつ大量にオートファゴソームに取り込んで分解するという特徴をもっている.
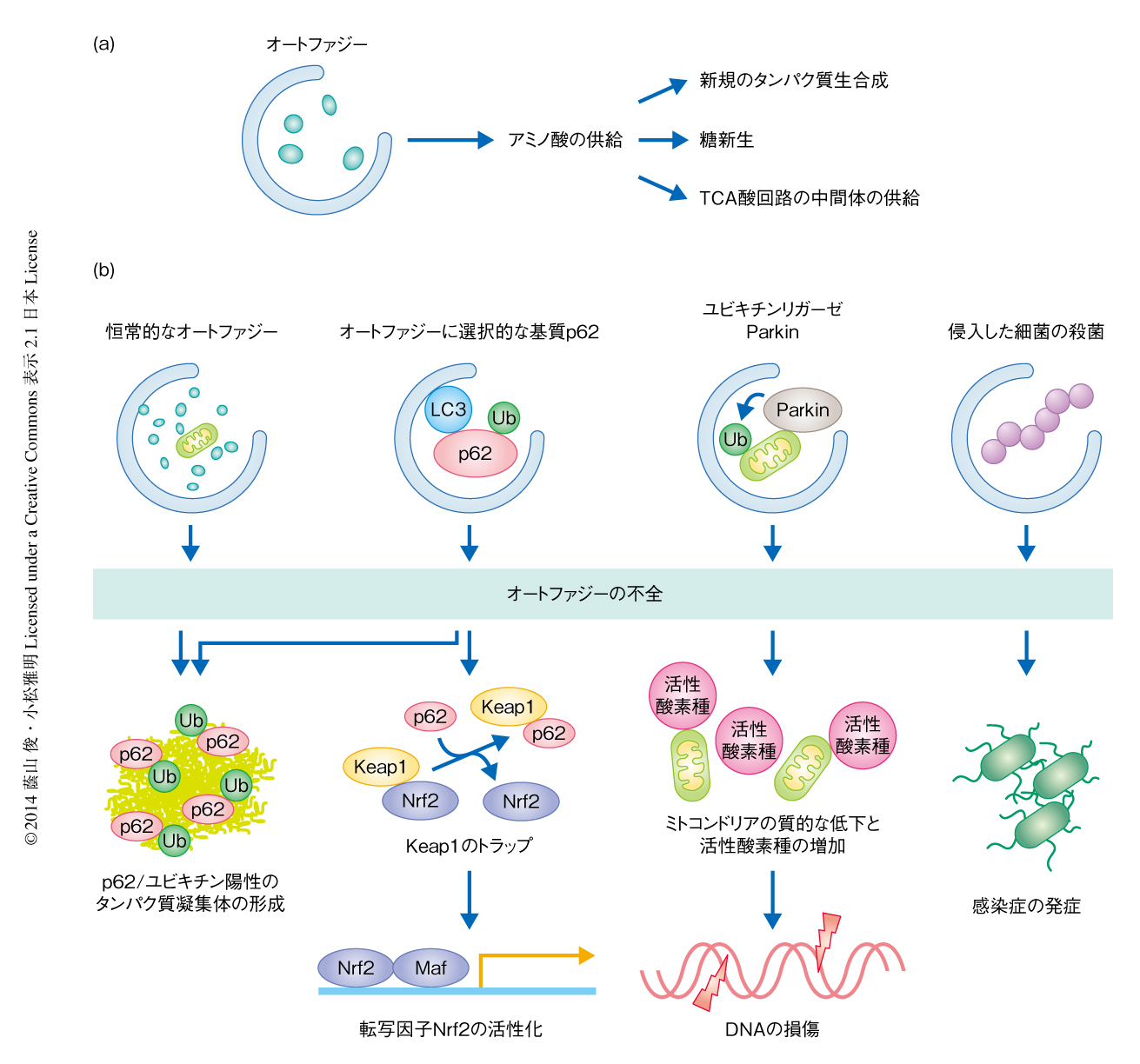
栄養が十分に存在する場合でも,基底レベルでの恒常的なオートファジーは起こっている(図3b).この恒常的なオートファジーの活性は低いレベルではあるが,細胞質のオルガネラやタンパク質の新陳代謝を担う.事実,一連の組織に特異的なオートファジー欠損マウスの解析により,障害をうけたミトコンドリアをはじめとした異常なオルガネラやユビキチンに陽性のタンパク質凝集体の蓄積がオートファジーの抑制により促進され,多様かつ重篤な疾患がひき起こされることが示された.オートファジーは細胞質のオルガネラやタンパク質を無作為に取り囲んだオートファゴソームがリソソームと融合して起こる非選択的な分解系と想定されてきたが,1990年代後半から2000年代前半,出芽酵母やメタノール資化性酵母を用いた解析から,それぞれ,アミノペプチダーゼ複合体やペルオキシソームがオートファジーの分子機構に依存して選択的に液胞(酵母において,リソソームに相当するオルガネラ)に輸送されていることが明らかになった27,28).2004年には,細胞に侵入したA型連鎖球菌がオートファゴソームに選択的に取り込まれリソソームにおいて殺菌されることが報告された29).2005年には,ユビキチンおよびLC3結合タンパク質SQSTM1/p62を介したオートファジーによるユビキチン化したタンパク質凝集体の排除機構が提唱された30).時期を同じくして,オートファジー欠損マウスの組織における共通の特徴として,Sqstm1/p62がいちじるしく蓄積し,ユビキチン陽性およびSqstm1/p62陽性のタンパク質凝集体が形成されていることが判明した31).また2009年には,出芽酵母においてミトコンドリアに対する選択的なオートファジーに必須のタンパク質としてAtg32が発見された32).さらに,若年性パーキンソン病の原因遺伝子にコードされるユビキチンリガーゼParkinによるミトコンドリア外膜タンパク質のユビキチン化が,オートファジーによる変性ミトコンドリアの除去を惹起することが報告されている33).これらの選択的オートファジーは細胞のストレスに応じて誘導的に起こる.この数年のあいだに,選択的オートファジーの異常はパーキンソン病,感染症,がんの発症に密接に関与することが明らかになってきた(図3b).
オートファジー関連遺伝子や選択的オートファジーに関与する遺伝子の変異により起こる遺伝性疾患,クローン病,パーキンソン病,SENDA病,Vici症候群,そして,がんについて述べる(表1).

クローン病は,消化管に重篤な炎症あるいは潰瘍をひき起こす原因不明の炎症性腸疾患である.クローン病の発症には,病原体の感染,自然免疫の異常,食物の成分による腸管粘膜の異常など,複雑な環境要因がからんでいると想定されている.2007年,コアAtgタンパク質をコードする遺伝子のひとつATG16L1遺伝子の非同義置換にクローン病との強い相関のあることが報告された34,35).ATG16L1はN末端側にコイルドコイル領域,中央からC末端にWDリピートをもつ.ATG12-ATG5複合体と相互作用して3者複合体を形成し,LC3共有結合系においてユビキチンリガーゼ様タンパク質として機能する36).見い出された非同義置換はWDリピートのN末端側に位置していた.このWDリピートは出芽酵母のATG16には存在せず,少なくとも,マウスの線維芽細胞ではオートファジーの活性に影響をあたえないことが報告されていた37,38).しかしごく最近,飢餓などの代謝ストレスのもとではこの変異型Atg16L1はカスパーゼ3による切断を容易にうけ,オートファジーの活性が減弱することが報告された.変異型Atg16ノックインマウスに腸内細菌のひとつであるYersinia属細菌を感染させると,オートファジーによる細菌の排除が行われず,炎症性サイトカインの放出が増加する39).このほかにも,Atg16L1を欠損したマクロファージはグラム陰性細菌に応答して過剰なインターロイキン1βおよびインターロイキン18を産生し強い炎症反応をひき起こすこと,Atg16L1を低発現するマウスでは抗菌ペプチドなどを分泌する小腸のPaneth細胞において分泌顆粒の形成不全および分泌異常が認められること,オートファジーと小胞体ストレス応答の両方を阻害することによりはじめて腸炎がひき起こされることなど,クローン病の発症にオートファジーの異常が関与するとする報告が蓄積しつつある40-42).
パーキンソン病は,神経伝達物質であるドーパミンを産生する中脳黒質のドーパミンニューロンの変性および脱落にともなう,線条体におけるドーパミンの含有量の減少に起因する.線条体におけるドーパミンの減少は大脳皮質前頭葉の運動野と大脳基底核をめぐるループ回路の機能失調をひき起こし,無動,固縮,振戦などの重篤な運動障害を発症する.パーキンソン病の大部分は弧発性であるが,特定の遺伝子変異に起因する家族性パーキンソン病もある.常染色体劣性遺伝性パーキンソン病の原因遺伝子であるPARK2遺伝子にコードされるユビキチンリガーゼParkinが,変性したミトコンドリアに転移しミトコンドリア外膜タンパク質をユビキチン化すると,それが引き金となりオートファジーによる異常なミトコンドリアの選択的な排除(マイトファジー)の惹起されることが報告された33)(図3b).このParkinのミトコンドリアへの局在は,常染色体劣性遺伝性パーキンソン病の別の原因遺伝子であるPARK6遺伝子にコードされるPINK1に依存する43-47).ミトコンドリア局在シグナルおよび膜貫通領域をもつセリン-スレオニンキナーゼであるPINK1は,通常はミトコンドリア内膜に存在するプロテアーゼPARLにより切断されプロテアソームにより分解される48,49).一方,膜電位を消失したミトコンドリアではPINK1はミトコンドリア外膜にとどまる.その結果,PINK1は自己リン酸化に依存してParkinをミトコンドリアへとリクルートする50).さらにごく最近,PINK1がユビキチンをリン酸化し,このユビキチンのリン酸化がParkinの活性化に不可欠であることが判明した51)(新着論文レビュー でも掲載).パーキンソン病の患者に由来する変異をもつParkinおよびPINK1はこれらの過程に異常を示す45).したがって,PINK-Parkin経路を介したマイトファジーの異常が活性酸素種の蓄積をひき起こし,ドーパミンニューロンが損傷してパーキンソン病の発症にいたると想定される.この仮説を支持するように,ミトコンドリアの異常とパーキンソン病との関連性は古くから報告されてきた.たとえば,パーキンソン病の患者において,ミトコンドリアDNAの欠失,呼吸活性の低下したミトコンドリアの蓄積,それにともなう活性酸素種の増加が確認されている.一方,PINK-Parkin経路はマイトファジーの誘導ではなく,おもにプロテアソームによるミトコンドリア外膜タンパク質の分解(ミトコンドリア外膜タンパク質の品質管理)に機能するとも考えられている52).PINK-Parkin経路が精力的に研究される一方,ミトコンドリアの分解までフォローした研究は少なく,今後の詳細な研究が必要と思われる.
SENDA病は,脳の黒質および淡蒼球における鉄の沈着と大脳の萎縮をともなう神経変性疾患である(SENDA:static encephalopathy of childhood with neurodegeneration in adulthood).最近,SENDA病の患者のエクソーム解析から,出芽酵母のオートファジー関連遺伝子であるATG18遺伝子のヒトにおける4つのホモログ(WIPI1遺伝子~WIPI4遺伝子)のひとつであるWDR45/WIPI4遺伝子に新規の変異が同定された53,54)(文献54) は,新着論文レビュー でも掲載).出芽酵母ATG18はPROPPINファミリーのひとつでβプロペラ構造をもつWD40ドメインを含み,ホスファチジルイノシトール-1,4,5-トリスリン酸(PI3P)およびリソソームに局在するホスファチジルイノシトール-3,5-ビスリン酸と結合する55-57).ATG18には2つのホスファチジルイノシトール結合部位が存在し,PI3キナーゼにより産生されたPI3Pに依存してオートファゴソームの形成される位置にリクルートされる58-61).SENDA病の患者から単離されたリンパ芽球様細胞株においてWDR45/WIPI4が顕著に減少しており,オートファゴソームの形成に関与するAtg9L1およびLC3の異常な蓄積(オートファゴソーム中間体の蓄積),および,オートファジーの活性の低下が確認された54).このことは,コアAtgタンパク質をコードするWDR45/WIPI4遺伝子における変異が神経変性疾患の発症因子となっていることを意味するが,オートファジーの障害がどのようにして鉄の沈着をひき起こすのか,なぜニューロンのみで影響が認められるかは不明であり,さらなる解析が必要である.WDR45/WIPI4遺伝子はX染色体に位置しており,X染色体の不活性化により女性のSENDA病の患者はWDR45/WIPI4の機能をモザイク状に失っている.男性ではWDR45/WIPI4は完全に欠損しているが,少数の男性例が報告されている53,62).WDR45/WIPI4の機能の欠損はWIPI1~WIPI3によりある程度は補完されると考えられ,わずかなオートファジーの活性の減弱が病態の発症に十分であることが示唆される.
Vici症候群は,劣性遺伝性の多系統疾患であり,脳梁の形成不全,白内障,心筋症,複合免疫不全,色素沈着の減少を特徴とする.Vici症候群の患者のエクソーム解析により,EPG5遺伝子に複数の変異を同定された63).EPG5遺伝子は線虫においてオートファジーの基質の分解阻害を指標としたスクリーニングにより後生動物に特異的なオートファジー関連遺伝子として同定された64).哺乳類細胞においてEPG5遺伝子をノックダウンすると電子密度の高いオートリソソームが長時間にわたり蓄積することから,EPG5はオートリソソームの成熟に関与すると考えられる65).実際に,Vici症候群の患者に由来する線維芽細胞および筋肉ではオートファジーの活性の低下とともに,オートファジーの選択的な基質であるSQSTM1/p62およびNBR1の蓄積が確認される.興味深いことに,Vici症候群の患者に由来する線維芽細胞ではオートファゴソームの形成を制御するAKTやFOXO3などの上流タンパク質も影響をうけていた63).したがって,Vici症候群ではオートファジーのさまざまなステップが影響をうけている可能性がある.さらに,EPG5ノックアウトマウスには大脳皮質の内錐体ニューロン層および脊髄の運動ニューロンに特異的な障害が確認され,筋肉の除神経,筋繊維の萎縮,生存率の低下,筋萎縮性側索硬化症様の症状を示す.EPG5遺伝子を欠損したマウスのニューロンおよびグリア細胞ではSqstm1/p62陽性タンパク質およびユビキチン化タンパク質も蓄積しており,オートファジーの活性の低下が認められた65).これらの事実は,EPG5遺伝子の変異によるVici症候群の発症にオートファジーの不全が関与することを強く示唆する.
オートファジーはインスリンシグナル伝達経路およびアミノ酸シグナル伝達経路の下流に位置するmTOR複合体1により負に制御されている(図2).インスリンシグナル伝達系に関与するがん遺伝子あるいはがん抑制遺伝子の変異はインスリンシグナルの恒常的な活性化をひき起こすことから,がん細胞におけるオートファジーの抑制が示唆されてきた.しかし,mTOR複合体1は多様な細胞機能を制御しているため,mTOR複合体1の活性化によるオートファジーの抑制がどこまで腫瘍の形成にかかわっているかは不明であった.1999年に,出芽酵母ATG6/VPS30遺伝子の高等動物におけるホモログであるBECN1遺伝子ががん抑制遺伝子であることが報告された66).BECN1はヒトの乳がんなどにおいて発現が低下しており,さらに,Becn1ヘテロノックアウトマウスではさまざまな腫瘍が自然発生することから,オートファジーは腫瘍の形成を抑制していると考えられてきた66).しかし,BECN1遺伝子のコードするBeclin 1はオートファジーだけではなくエンドサイトーシスにも関与する多機能タンパク質であるため,Becn1ヘテロノックアウトマウスを用いた実験の結果が真にオートファジー不全の表現型を示しているのか疑問が残っていた.
2011年に,オートファジーに特化した遺伝子改変マウスが腫瘍を形成することが判明し,腫瘍の抑制がオートファジーの生理的な意義のひとつとして確固たるものになった.Atg5遺伝子を全身においてモザイク状に欠損させたマウスは,生後約7カ月で肝臓に小さな腫瘍が観察され,加齢とともに腫瘍の大きさおよび数は増加し,約16カ月齢において肝臓は腫瘍によりしめられた67,68).興味深いことに,このモザイクマウスは全身においてAtg5遺伝子を欠損しているにもかかわらず,肝臓にのみ腫瘍が形成された68).ほぼ同様の結果が,肝臓に特異的なAtg7ノックアウトマウスにおいても観察されている67).注目すべき点として,Becn1ヘテロノックアウトマウスとは異なり,Atg5遺伝子あるいはAtg7遺伝子の欠損により形成された肝臓の腫瘍細胞には核の異数性がなく,発生部位に限局してモノクローナルに増殖し規則的な配列や増殖パターンを形成する良性の腫瘍であり,ほかの臓器への転移も確認されなかった67,68).Atg5遺伝子あるいはAtg7遺伝子を欠損した肝実質細胞において,膨化したミトコンドリアやペルオキシソームの蓄積,DNAの酸化損傷やゲノムの不安定性が認められる67,68).一定に保たれるべきペルオキシソームや機能不全におちいったミトコンドリアがオートファジーにより分解されないために細胞に蓄積し,その結果として活性酸素種が過剰に生じ,ついでDNA損傷が誘発され腫瘍の形成を促進している可能性がある.
Sqstm1/p62も腫瘍化および腫瘍の進行に関与する.Sqstm1/p62はオートファゴソームに局在するタンパク質であるLC3に結合するタンパク質であり,オートファジーにより恒常的に分解されている30).したがって,オートファジーを欠損した組織において顕著に蓄積する.ごく最近,腎細胞がんの患者の5%においてSQSTM1/p62遺伝子を含む領域が遺伝子重複していること,SQSTM1/p62の発現上昇が腫瘍の形成と相関することが報告された69).つまり,SQSTM1/p62はがん遺伝子であった.SQSTM1/p62は腫瘍化あるいは腫瘍の進行にどのように関与するのだろうか? Sqstm1/p62は複数のシグナル伝達タンパク質と相互作用するスカフォールドタンパク質であり,TRAF6との結合を介したNF-κBシグナルの活性化やユビキチン化したカスパーゼ-8との相互作用を介したアポトーシスの促進など細胞の生死を制御している70).NF-κBシグナルの活性化は腫瘍の形成に関与することから,Sqstm1/p62の蓄積によるNF-κBシグナルの恒常的な活性化が腫瘍の形成の要因としてあげられる.事実,SQSTM1/p62の欠失によるNF-κBシグナルの抑制がRas誘導性の肺腺がんの形成を抑制することや,恒常的なKrasの活性化によるSQSTM1/p62の発現の亢進が膵管腺がんの発達に関与することが報告されている71,72).
Sqstm1/p62はCullin3型ユビキチンリガーゼのアダプタータンパク質Keap1との相互作用を介して転写因子Nrf2を活性化し,ストレス応答性の遺伝子発現を誘導する31)(図3b).このKeap1との結合はmTOR複合体1によるSqstm1/p62のリン酸化により増強される73)(新着論文レビュー でも掲載).重要なことに,肺線がんなどにおいて恒常的にNrf2を活性化しているがん細胞は,ストレス応答性遺伝子のみならず,ペントースリン酸経路-プリンヌクレオチド合成経路やグルタミン代謝を制御する酵素の遺伝子発現も誘導し,これらが腫瘍の増殖をささえている74)(新着論文レビュー でも掲載).Sqstm1/p62が蓄積するとNrf2は恒常的に活性化され,その結果,腫瘍細胞の増殖を促進すると考えられる.このことは,肝臓に特異的なAtg7 Sqstm1/p62ダブルノックアウトマウスにおいて,腫瘍の形成が大幅に抑制されることと一致する.これらの事実は,オートファジーの欠損にともなうペルオキシソームや機能不全におちいったミトコンドリアの蓄積による活性酸素種の増加,および,Sqstm1/p62の蓄積によるシグナル伝達の異常が,腫瘍の形成および進行の原因であることを強く示唆する.
いちじるしく増殖をくり返すがん細胞は十分な血管新生がともなわず,低酸素あるいは低グルコースといった代謝ストレスのもとでの生存を余儀なくされている.また,がん細胞はその増殖を維持するため,アミノ酸,糖質,脂肪酸などをより多く必要とする,すなわち,がん細胞は代謝の要求性がきわめて高い状態にある75).オートファジーは腫瘍の形成を抑制する一方,いったん腫瘍化してしまった細胞においては栄養源を供給することにより代謝の要求性をみたしその増殖に貢献している.とくに,約90%にRas遺伝子の変異の認められる膵がんや活性型Rasにより形質転換したがん細胞などでは,オートファジーが恒常的に亢進した“オートファジー依存”の状態になっている25,76).このオートファジーの活性化の分子機構はほとんどわかっていないが,オートファジーを阻害すると,膵がんの増殖抑制,異種移植による腫瘍形成の抑制,膵がんマウスモデルの腫瘍の退縮がみられる25,76).このようなオートファジーの阻害による腫瘍の抑制効果は,Myc誘導性のリンパ腫やポリオーマウイルス中型T抗原に誘導性の乳腺腫瘍においても認められる77,78).
オートファジーを阻害した膵がん細胞では,活性酸素種の増加,DNA損傷の蓄積,くわえて,おそらくTCA回路中間体の供給の減少による酸化的リン酸化の低下が観察される25).したがって,これらのがん細胞において恒常的に亢進しているオートファジーは,アミノ酸をたえず供給することによりTCA回路中間体の産生を促進するとともに,ミトコンドリアの品質管理によりがん細胞の生存および増殖に寄与していると想定される.しかし,Ras遺伝子のオートファジーへの関与は複雑である.たとえば,Rasの過剰発現により誘導されるオートファジーは,ヒトの卵巣表層上皮細胞においては細胞死を,線維芽細胞においては細胞老化を促進する76,79,80).また,一般的にRASの活性化が確認される黒色腫においては,mTOR複合体1の恒常的な活性化によりオートファジーが抑制されているらしい.したがって,黒色腫はアミノ酸プールの減少に応答したオートファジーを誘導できず細胞死をひき起こす81).種々のがんにおけるオートファジーの生理的な役割は,環境やステージ,そして,遺伝学的な背景により異なるのだろう.事実,最近,p53の欠失の有無により膵がんにおけるオートファジーの役割が異なることが明らかになった82).
オートファジー関連遺伝子の同定をはじめ,オートファジー研究分野における日本人の貢献は大きい.しかし現在,欧米およびアジアの各国ではオートファジーの研究において基礎レベルのみならずトランスレーショナルリサーチも活発に行われており,臨床への応用に関してわが国は後塵を拝している感がある.とくに,がん細胞においてはオートファジーによるアミノ酸の供給やミトコンドリアなどのオルガネラの品質管理がその増殖に決定的であり,既存の抗がん剤とオートファジー阻害剤(正確には,リソソーム酸性化阻害剤)をあわせたがん治療の臨床試験がすでに欧米において開始されている.また,世界中でオートファジーに特異的な活性化剤および阻害剤のスクリーニングが行われ,いくつかは動物レベルでその効果が確認されている.しかし,オートファジーの臨床への応用にはいくつかの問題点が残っている.オートファジーの分子機構には未解明の謎がいまだ山積しており,その阻害あるいは亢進によりどのような副作用があるのかも不透明である.現在まで,オートファジーの活性をモニターできるマーカーもプローブも存在せず,薬効の判定ができない.遺伝子改変動物の解析によりおのおのの組織におけるオートファジーの生理機能の大筋は判明しつつあるが,ヒトの病態におけるその意義はほとんど手つかずの状態であり,さらなる解析が必要であろう.
略歴:2011年 大阪大学大学院生命機能研究科 修了,同年 東京都医学総合研究所 研究員を経て,2014年より新潟大学大学院医歯学総合研究科 助教.
研究テーマ:生体におけるオートファジーの生理的な意義および分子機構.とくに,選択的オートファジーに着目し,疾患とのかかわりを明らかにしていきたい.
小松 雅明(Masaaki Komatsu)
新潟大学大学院医歯学総合研究科 教授.
研究室URL:http://www.med.niigata-u.ac.jp/bc1/welcome.html
© 2014 蔭山 俊・小松雅明 Licensed under CC 表示 2.1 日本
(新潟大学大学院医歯学総合研究科 分子生物学)
email:小松雅明
領域融合レビュー, 3, e006 (2014) DOI: 10.7875/leading.author.3.e006
Shun Kageyama & Masaaki Komatsu: Autophagy and disease states.
要 約
オートファジー関連遺伝子が同定されて以来,さまざまなモデル動物においてオートファジー関連遺伝子の改変動物が作出され,オートファジーの生理機能,とくに,アミノ酸プールの維持や細胞の新陳代謝としての機能が明確になった.また,さまざまな臓器に特異的なオートファジー関連遺伝子の改変マウスが,変性タンパク質そして異常なオルガネラの蓄積をともなう神経変性疾患,肝障害,がんなど重篤な疾患を呈することが明らかになり,オートファジーによる細胞保護の作用が決定的になった.さらに,細胞に侵入した細菌や異常なオルガネラ,とくに変性ミトコンドリアのオートファジーによる選択的な除去機構が明らかにされ,その病態生理的な役割は複雑さを増している.そしてごく最近,ヒトにおいてもオートファジー関連遺伝子の変異が神経変性疾患や炎症性疾患をひき起こすことが発見され,オートファジーの異常がヒトの疾患にも直接に関与することが判明した.このレビューではオートファジーを概説するとともに,オートファジー関連遺伝子の変異により起こる遺伝性疾患,オートファジーの異常が複雑に関与するがんをとりあげる.
はじめに
オートファジーはリソソームにおいて細胞質の成分を分解する機構である.オートファジーにはミクロオートファジー,シャペロン介在性オートファジー,マクロオートファジーの3つの経路がある.これらのなかで,マクロオートファジーの解析がもっとも進んでおり,たんに“オートファジー”と表記した場合にはマクロオートファジーをさすことが多く,このレビューにおいてもこれにしたがう.オートファジーは細胞質に出現した隔離膜が細胞質の成分を取り囲みオートファゴソームが形成される過程と,オートファゴソームがリソソームと融合し細胞質の成分を分解する過程からなる.小胞体,ミトコンドリア,ゴルジ体,細胞膜,さらには,リサイクリングエンドソームに由来する多彩な膜成分がオートファゴソーム膜の形成に寄与しており,その形成から内容物の分解までは,オートファジー関連遺伝子(autophagy-related gene:ATG)にコードされるAtgタンパク質により厳密に制御されている.
オートファゴソームの形成に特化したコアAtgタンパク質は,以下の6つの機能単位に分類される.1)ULK1キナーゼ複合体(ULK1,ULK2,Atg13,FIP200,Atg101),2)クラスIIIホスファチジルイノシトール3-キナーゼ(PI3キナーゼ)複合体(Vps34,Vps15,Beclin 1,Atg14(L)/Barkor),3)ホスファチジルイノシトール-1,4,5-トリスリン酸(PI3P)結合タンパク質(Atg2,WIPI1~WIPI4),4)膜タンパク質(Atg9L1),5)Atg12共有結合系(Atg12,Atg7,Atg10,Atg5,Atg16L1,Atg16L2),6)LC3共有結合系(LC3A~LC3C,GABARAP,GABARAPL1,GABARAPL2,Atg4A~Atg4D,Atg7,Atg3).これらコアAtgタンパク質の機能単位が連続的かつ協調的に作動することによりオートファゴソームは形成される1)(図1).
なお,出芽酵母におけるオートファジーの研究については,荒木保弘・大隅良典, 領域融合レビュー, 1, e005, 2012 も参照されたい.
1.オートファジーの活性化および抑制の機構
オートファジーはおもにインスリンシグナルおよびアミノ酸シグナルに応じたmTOR複合体1の活性化により抑制される2-4).インスリンシグナルはクラスI PI3キナーゼ-AKT経路を活性化し,TSC2をリン酸化することによりその活性を抑制する.TSC2はGTPase活性化タンパク質(GTPase activating protein:GAP)として低分子量Gタンパク質であるRhebの活性を負に制御する5-8).したがって,AKTによるTSC2の抑制はmTOR複合体1の活性化を促進する(図2).一方,アミノ酸シグナル(とくに,ロイシンおよびグルタミン)により低分子量Gタンパク質であるRagがGDP結合型からGTP結合型へと変換されると,RagはmTOR複合体1の構成タンパク質であるRaptorと結合する.さらに,Ragはリソソーム膜に局在するRagulatorとよばれる複合体と相互作用することによりmTOR複合体1をリソソーム膜につなぎとめる.最終的に,リソソーム膜に存在するRhebとmTOR複合体1とが会合することによりmTOR複合体1の活性化が促進される9-11).ロイシルtRNAシンテターゼはRagのGTPase活性化タンパク質として機能し,ロイシンに誘導されるmTOR複合体1の活性化に寄与する.グルタミンはグルタミナーゼによりグルタミン酸に変換され,さらに,グルタミン酸デヒドロゲナーゼによる脱アミノ化によりαケトグルタル酸とアンモニアになる.αケトグルタル酸はRagのGTP結合型への変換を促進しmTOR複合体1を活性化する12-14).このとき,ロイシンはグルタミン酸デヒドロゲナーゼをアロステリックに活性化している.他方,リソソームに存在するアミノ酸はリソソーム膜に局在するV型ATPaseが感知し,V型ATPaseとRagulatorとの会合あるいは解離を制御することによりmTOR複合体1の活性を制御する15,16).活性化したmTOR複合体1はULK1キナーゼ複合体と相互作用し,ULK1およびAtg13がリン酸化されオートファジーは抑制される17)(図2).すなわち,インスリンあるいはアミノ酸が欠乏するとオートファジーは容易に惹起される(図2).骨格筋ではインスリンにより,一方,肝臓ではアミノ酸によりオートファジーが抑制されており,生体においてオートファジーはmTOR複合体1により組織ごとに異なった分子機構により制御されている18).グルコースが欠乏するとATPが減少してAMP/ATP比が上昇し,その結果,AMPKが活性化する.AMPKはRaptorのリン酸化あるいはTSC2のリン酸化を介してmTOR複合体1を不活性化させオートファジーを誘導する19,20).また,AMPKはULK1およびBeclin 1を直接にリン酸化し活性化する21,22).したがって,AMPKはmTOR複合体1に依存的にも非依存的にオートファジーを活性化しうる(図2).
2.オートファジーの生理機能
オートファジーのもっとも基本的な生理機能は,飢餓への適応である(図3a).すでに述べたとおり,オートファジーは低アミノ酸状態あるいは低インスリン状態において一過的に誘導され細胞質の成分を分解する.糖や脂質と異なりアミノ酸プールはわずかであり(ヒトの成人の場合,タンパク質生合成に必要なアミノ酸量は1日あたり300~400 gであるのに対し,アミノ酸プールは約100 gしかない),飢餓に応答したオートファジーによるアミノ酸の供給が重要であることは容易に想像できる.たとえば,マウスの新生仔は胎盤からの栄養の供給が絶たれる出生の直後に一過的にオートファジーを誘導しアミノ酸プールを維持する.全身においてAtg5遺伝子を欠損させたマウスは出生の直後の飢餓の際に組織および血中において重篤な低アミノ酸症状におちいり,それが一因となり出生から24時間以内に死亡する23).では,オートファジーにより生じたアミノ酸はどうなるのだろうか? 出芽酵母のオートファジー不能変異体は飢餓の際にミトコンドリア呼吸鎖タンパク質や抗酸化タンパク質の生合成の不全をひき起こすことから,第1に,飢餓に応答した特定のタンパク質の生合成のために利用されると考えられる.また,アミノ酸はアミノ基転移反応やアミノ酸代謝によりピルビン酸,TCA回路中間体,アセチルCoAとなる.アミノ酸はこの補充反応経路により糖新生,脂肪酸生合成,エネルギー産生に寄与する.実際に,肝臓に特異的なAtg7ノックアウトマウスの解析から,オートファジーにより生じたアミノ酸は糖新生に利用されることが明らかになっている24).がん細胞ではオートファジーに依存したアミノ酸およびTCA回路中間体の供給がその生存に重要らしい25).また,Atg5ノックアウトマウスにおいてはAMPKが活性化されており,糖新生が阻害されていることが示唆される23).受精の直後にもオートファジーは活性化され,母方に由来するタンパク質を分解し,胚の初期発生に必要なタンパク質の生合成のためのアミノ酸を供給する26).このようなオートファジーは,細胞質の成分を非選択的かつ大量にオートファゴソームに取り込んで分解するという特徴をもっている.
栄養が十分に存在する場合でも,基底レベルでの恒常的なオートファジーは起こっている(図3b).この恒常的なオートファジーの活性は低いレベルではあるが,細胞質のオルガネラやタンパク質の新陳代謝を担う.事実,一連の組織に特異的なオートファジー欠損マウスの解析により,障害をうけたミトコンドリアをはじめとした異常なオルガネラやユビキチンに陽性のタンパク質凝集体の蓄積がオートファジーの抑制により促進され,多様かつ重篤な疾患がひき起こされることが示された.オートファジーは細胞質のオルガネラやタンパク質を無作為に取り囲んだオートファゴソームがリソソームと融合して起こる非選択的な分解系と想定されてきたが,1990年代後半から2000年代前半,出芽酵母やメタノール資化性酵母を用いた解析から,それぞれ,アミノペプチダーゼ複合体やペルオキシソームがオートファジーの分子機構に依存して選択的に液胞(酵母において,リソソームに相当するオルガネラ)に輸送されていることが明らかになった27,28).2004年には,細胞に侵入したA型連鎖球菌がオートファゴソームに選択的に取り込まれリソソームにおいて殺菌されることが報告された29).2005年には,ユビキチンおよびLC3結合タンパク質SQSTM1/p62を介したオートファジーによるユビキチン化したタンパク質凝集体の排除機構が提唱された30).時期を同じくして,オートファジー欠損マウスの組織における共通の特徴として,Sqstm1/p62がいちじるしく蓄積し,ユビキチン陽性およびSqstm1/p62陽性のタンパク質凝集体が形成されていることが判明した31).また2009年には,出芽酵母においてミトコンドリアに対する選択的なオートファジーに必須のタンパク質としてAtg32が発見された32).さらに,若年性パーキンソン病の原因遺伝子にコードされるユビキチンリガーゼParkinによるミトコンドリア外膜タンパク質のユビキチン化が,オートファジーによる変性ミトコンドリアの除去を惹起することが報告されている33).これらの選択的オートファジーは細胞のストレスに応じて誘導的に起こる.この数年のあいだに,選択的オートファジーの異常はパーキンソン病,感染症,がんの発症に密接に関与することが明らかになってきた(図3b).
3.オートファジーと疾患
オートファジー関連遺伝子や選択的オートファジーに関与する遺伝子の変異により起こる遺伝性疾患,クローン病,パーキンソン病,SENDA病,Vici症候群,そして,がんについて述べる(表1).
クローン病は,消化管に重篤な炎症あるいは潰瘍をひき起こす原因不明の炎症性腸疾患である.クローン病の発症には,病原体の感染,自然免疫の異常,食物の成分による腸管粘膜の異常など,複雑な環境要因がからんでいると想定されている.2007年,コアAtgタンパク質をコードする遺伝子のひとつATG16L1遺伝子の非同義置換にクローン病との強い相関のあることが報告された34,35).ATG16L1はN末端側にコイルドコイル領域,中央からC末端にWDリピートをもつ.ATG12-ATG5複合体と相互作用して3者複合体を形成し,LC3共有結合系においてユビキチンリガーゼ様タンパク質として機能する36).見い出された非同義置換はWDリピートのN末端側に位置していた.このWDリピートは出芽酵母のATG16には存在せず,少なくとも,マウスの線維芽細胞ではオートファジーの活性に影響をあたえないことが報告されていた37,38).しかしごく最近,飢餓などの代謝ストレスのもとではこの変異型Atg16L1はカスパーゼ3による切断を容易にうけ,オートファジーの活性が減弱することが報告された.変異型Atg16ノックインマウスに腸内細菌のひとつであるYersinia属細菌を感染させると,オートファジーによる細菌の排除が行われず,炎症性サイトカインの放出が増加する39).このほかにも,Atg16L1を欠損したマクロファージはグラム陰性細菌に応答して過剰なインターロイキン1βおよびインターロイキン18を産生し強い炎症反応をひき起こすこと,Atg16L1を低発現するマウスでは抗菌ペプチドなどを分泌する小腸のPaneth細胞において分泌顆粒の形成不全および分泌異常が認められること,オートファジーと小胞体ストレス応答の両方を阻害することによりはじめて腸炎がひき起こされることなど,クローン病の発症にオートファジーの異常が関与するとする報告が蓄積しつつある40-42).
パーキンソン病は,神経伝達物質であるドーパミンを産生する中脳黒質のドーパミンニューロンの変性および脱落にともなう,線条体におけるドーパミンの含有量の減少に起因する.線条体におけるドーパミンの減少は大脳皮質前頭葉の運動野と大脳基底核をめぐるループ回路の機能失調をひき起こし,無動,固縮,振戦などの重篤な運動障害を発症する.パーキンソン病の大部分は弧発性であるが,特定の遺伝子変異に起因する家族性パーキンソン病もある.常染色体劣性遺伝性パーキンソン病の原因遺伝子であるPARK2遺伝子にコードされるユビキチンリガーゼParkinが,変性したミトコンドリアに転移しミトコンドリア外膜タンパク質をユビキチン化すると,それが引き金となりオートファジーによる異常なミトコンドリアの選択的な排除(マイトファジー)の惹起されることが報告された33)(図3b).このParkinのミトコンドリアへの局在は,常染色体劣性遺伝性パーキンソン病の別の原因遺伝子であるPARK6遺伝子にコードされるPINK1に依存する43-47).ミトコンドリア局在シグナルおよび膜貫通領域をもつセリン-スレオニンキナーゼであるPINK1は,通常はミトコンドリア内膜に存在するプロテアーゼPARLにより切断されプロテアソームにより分解される48,49).一方,膜電位を消失したミトコンドリアではPINK1はミトコンドリア外膜にとどまる.その結果,PINK1は自己リン酸化に依存してParkinをミトコンドリアへとリクルートする50).さらにごく最近,PINK1がユビキチンをリン酸化し,このユビキチンのリン酸化がParkinの活性化に不可欠であることが判明した51)(新着論文レビュー でも掲載).パーキンソン病の患者に由来する変異をもつParkinおよびPINK1はこれらの過程に異常を示す45).したがって,PINK-Parkin経路を介したマイトファジーの異常が活性酸素種の蓄積をひき起こし,ドーパミンニューロンが損傷してパーキンソン病の発症にいたると想定される.この仮説を支持するように,ミトコンドリアの異常とパーキンソン病との関連性は古くから報告されてきた.たとえば,パーキンソン病の患者において,ミトコンドリアDNAの欠失,呼吸活性の低下したミトコンドリアの蓄積,それにともなう活性酸素種の増加が確認されている.一方,PINK-Parkin経路はマイトファジーの誘導ではなく,おもにプロテアソームによるミトコンドリア外膜タンパク質の分解(ミトコンドリア外膜タンパク質の品質管理)に機能するとも考えられている52).PINK-Parkin経路が精力的に研究される一方,ミトコンドリアの分解までフォローした研究は少なく,今後の詳細な研究が必要と思われる.
SENDA病は,脳の黒質および淡蒼球における鉄の沈着と大脳の萎縮をともなう神経変性疾患である(SENDA:static encephalopathy of childhood with neurodegeneration in adulthood).最近,SENDA病の患者のエクソーム解析から,出芽酵母のオートファジー関連遺伝子であるATG18遺伝子のヒトにおける4つのホモログ(WIPI1遺伝子~WIPI4遺伝子)のひとつであるWDR45/WIPI4遺伝子に新規の変異が同定された53,54)(文献54) は,新着論文レビュー でも掲載).出芽酵母ATG18はPROPPINファミリーのひとつでβプロペラ構造をもつWD40ドメインを含み,ホスファチジルイノシトール-1,4,5-トリスリン酸(PI3P)およびリソソームに局在するホスファチジルイノシトール-3,5-ビスリン酸と結合する55-57).ATG18には2つのホスファチジルイノシトール結合部位が存在し,PI3キナーゼにより産生されたPI3Pに依存してオートファゴソームの形成される位置にリクルートされる58-61).SENDA病の患者から単離されたリンパ芽球様細胞株においてWDR45/WIPI4が顕著に減少しており,オートファゴソームの形成に関与するAtg9L1およびLC3の異常な蓄積(オートファゴソーム中間体の蓄積),および,オートファジーの活性の低下が確認された54).このことは,コアAtgタンパク質をコードするWDR45/WIPI4遺伝子における変異が神経変性疾患の発症因子となっていることを意味するが,オートファジーの障害がどのようにして鉄の沈着をひき起こすのか,なぜニューロンのみで影響が認められるかは不明であり,さらなる解析が必要である.WDR45/WIPI4遺伝子はX染色体に位置しており,X染色体の不活性化により女性のSENDA病の患者はWDR45/WIPI4の機能をモザイク状に失っている.男性ではWDR45/WIPI4は完全に欠損しているが,少数の男性例が報告されている53,62).WDR45/WIPI4の機能の欠損はWIPI1~WIPI3によりある程度は補完されると考えられ,わずかなオートファジーの活性の減弱が病態の発症に十分であることが示唆される.
Vici症候群は,劣性遺伝性の多系統疾患であり,脳梁の形成不全,白内障,心筋症,複合免疫不全,色素沈着の減少を特徴とする.Vici症候群の患者のエクソーム解析により,EPG5遺伝子に複数の変異を同定された63).EPG5遺伝子は線虫においてオートファジーの基質の分解阻害を指標としたスクリーニングにより後生動物に特異的なオートファジー関連遺伝子として同定された64).哺乳類細胞においてEPG5遺伝子をノックダウンすると電子密度の高いオートリソソームが長時間にわたり蓄積することから,EPG5はオートリソソームの成熟に関与すると考えられる65).実際に,Vici症候群の患者に由来する線維芽細胞および筋肉ではオートファジーの活性の低下とともに,オートファジーの選択的な基質であるSQSTM1/p62およびNBR1の蓄積が確認される.興味深いことに,Vici症候群の患者に由来する線維芽細胞ではオートファゴソームの形成を制御するAKTやFOXO3などの上流タンパク質も影響をうけていた63).したがって,Vici症候群ではオートファジーのさまざまなステップが影響をうけている可能性がある.さらに,EPG5ノックアウトマウスには大脳皮質の内錐体ニューロン層および脊髄の運動ニューロンに特異的な障害が確認され,筋肉の除神経,筋繊維の萎縮,生存率の低下,筋萎縮性側索硬化症様の症状を示す.EPG5遺伝子を欠損したマウスのニューロンおよびグリア細胞ではSqstm1/p62陽性タンパク質およびユビキチン化タンパク質も蓄積しており,オートファジーの活性の低下が認められた65).これらの事実は,EPG5遺伝子の変異によるVici症候群の発症にオートファジーの不全が関与することを強く示唆する.
4.オートファジーとがん
オートファジーはインスリンシグナル伝達経路およびアミノ酸シグナル伝達経路の下流に位置するmTOR複合体1により負に制御されている(図2).インスリンシグナル伝達系に関与するがん遺伝子あるいはがん抑制遺伝子の変異はインスリンシグナルの恒常的な活性化をひき起こすことから,がん細胞におけるオートファジーの抑制が示唆されてきた.しかし,mTOR複合体1は多様な細胞機能を制御しているため,mTOR複合体1の活性化によるオートファジーの抑制がどこまで腫瘍の形成にかかわっているかは不明であった.1999年に,出芽酵母ATG6/VPS30遺伝子の高等動物におけるホモログであるBECN1遺伝子ががん抑制遺伝子であることが報告された66).BECN1はヒトの乳がんなどにおいて発現が低下しており,さらに,Becn1ヘテロノックアウトマウスではさまざまな腫瘍が自然発生することから,オートファジーは腫瘍の形成を抑制していると考えられてきた66).しかし,BECN1遺伝子のコードするBeclin 1はオートファジーだけではなくエンドサイトーシスにも関与する多機能タンパク質であるため,Becn1ヘテロノックアウトマウスを用いた実験の結果が真にオートファジー不全の表現型を示しているのか疑問が残っていた.
2011年に,オートファジーに特化した遺伝子改変マウスが腫瘍を形成することが判明し,腫瘍の抑制がオートファジーの生理的な意義のひとつとして確固たるものになった.Atg5遺伝子を全身においてモザイク状に欠損させたマウスは,生後約7カ月で肝臓に小さな腫瘍が観察され,加齢とともに腫瘍の大きさおよび数は増加し,約16カ月齢において肝臓は腫瘍によりしめられた67,68).興味深いことに,このモザイクマウスは全身においてAtg5遺伝子を欠損しているにもかかわらず,肝臓にのみ腫瘍が形成された68).ほぼ同様の結果が,肝臓に特異的なAtg7ノックアウトマウスにおいても観察されている67).注目すべき点として,Becn1ヘテロノックアウトマウスとは異なり,Atg5遺伝子あるいはAtg7遺伝子の欠損により形成された肝臓の腫瘍細胞には核の異数性がなく,発生部位に限局してモノクローナルに増殖し規則的な配列や増殖パターンを形成する良性の腫瘍であり,ほかの臓器への転移も確認されなかった67,68).Atg5遺伝子あるいはAtg7遺伝子を欠損した肝実質細胞において,膨化したミトコンドリアやペルオキシソームの蓄積,DNAの酸化損傷やゲノムの不安定性が認められる67,68).一定に保たれるべきペルオキシソームや機能不全におちいったミトコンドリアがオートファジーにより分解されないために細胞に蓄積し,その結果として活性酸素種が過剰に生じ,ついでDNA損傷が誘発され腫瘍の形成を促進している可能性がある.
Sqstm1/p62も腫瘍化および腫瘍の進行に関与する.Sqstm1/p62はオートファゴソームに局在するタンパク質であるLC3に結合するタンパク質であり,オートファジーにより恒常的に分解されている30).したがって,オートファジーを欠損した組織において顕著に蓄積する.ごく最近,腎細胞がんの患者の5%においてSQSTM1/p62遺伝子を含む領域が遺伝子重複していること,SQSTM1/p62の発現上昇が腫瘍の形成と相関することが報告された69).つまり,SQSTM1/p62はがん遺伝子であった.SQSTM1/p62は腫瘍化あるいは腫瘍の進行にどのように関与するのだろうか? Sqstm1/p62は複数のシグナル伝達タンパク質と相互作用するスカフォールドタンパク質であり,TRAF6との結合を介したNF-κBシグナルの活性化やユビキチン化したカスパーゼ-8との相互作用を介したアポトーシスの促進など細胞の生死を制御している70).NF-κBシグナルの活性化は腫瘍の形成に関与することから,Sqstm1/p62の蓄積によるNF-κBシグナルの恒常的な活性化が腫瘍の形成の要因としてあげられる.事実,SQSTM1/p62の欠失によるNF-κBシグナルの抑制がRas誘導性の肺腺がんの形成を抑制することや,恒常的なKrasの活性化によるSQSTM1/p62の発現の亢進が膵管腺がんの発達に関与することが報告されている71,72).
Sqstm1/p62はCullin3型ユビキチンリガーゼのアダプタータンパク質Keap1との相互作用を介して転写因子Nrf2を活性化し,ストレス応答性の遺伝子発現を誘導する31)(図3b).このKeap1との結合はmTOR複合体1によるSqstm1/p62のリン酸化により増強される73)(新着論文レビュー でも掲載).重要なことに,肺線がんなどにおいて恒常的にNrf2を活性化しているがん細胞は,ストレス応答性遺伝子のみならず,ペントースリン酸経路-プリンヌクレオチド合成経路やグルタミン代謝を制御する酵素の遺伝子発現も誘導し,これらが腫瘍の増殖をささえている74)(新着論文レビュー でも掲載).Sqstm1/p62が蓄積するとNrf2は恒常的に活性化され,その結果,腫瘍細胞の増殖を促進すると考えられる.このことは,肝臓に特異的なAtg7 Sqstm1/p62ダブルノックアウトマウスにおいて,腫瘍の形成が大幅に抑制されることと一致する.これらの事実は,オートファジーの欠損にともなうペルオキシソームや機能不全におちいったミトコンドリアの蓄積による活性酸素種の増加,および,Sqstm1/p62の蓄積によるシグナル伝達の異常が,腫瘍の形成および進行の原因であることを強く示唆する.
いちじるしく増殖をくり返すがん細胞は十分な血管新生がともなわず,低酸素あるいは低グルコースといった代謝ストレスのもとでの生存を余儀なくされている.また,がん細胞はその増殖を維持するため,アミノ酸,糖質,脂肪酸などをより多く必要とする,すなわち,がん細胞は代謝の要求性がきわめて高い状態にある75).オートファジーは腫瘍の形成を抑制する一方,いったん腫瘍化してしまった細胞においては栄養源を供給することにより代謝の要求性をみたしその増殖に貢献している.とくに,約90%にRas遺伝子の変異の認められる膵がんや活性型Rasにより形質転換したがん細胞などでは,オートファジーが恒常的に亢進した“オートファジー依存”の状態になっている25,76).このオートファジーの活性化の分子機構はほとんどわかっていないが,オートファジーを阻害すると,膵がんの増殖抑制,異種移植による腫瘍形成の抑制,膵がんマウスモデルの腫瘍の退縮がみられる25,76).このようなオートファジーの阻害による腫瘍の抑制効果は,Myc誘導性のリンパ腫やポリオーマウイルス中型T抗原に誘導性の乳腺腫瘍においても認められる77,78).
オートファジーを阻害した膵がん細胞では,活性酸素種の増加,DNA損傷の蓄積,くわえて,おそらくTCA回路中間体の供給の減少による酸化的リン酸化の低下が観察される25).したがって,これらのがん細胞において恒常的に亢進しているオートファジーは,アミノ酸をたえず供給することによりTCA回路中間体の産生を促進するとともに,ミトコンドリアの品質管理によりがん細胞の生存および増殖に寄与していると想定される.しかし,Ras遺伝子のオートファジーへの関与は複雑である.たとえば,Rasの過剰発現により誘導されるオートファジーは,ヒトの卵巣表層上皮細胞においては細胞死を,線維芽細胞においては細胞老化を促進する76,79,80).また,一般的にRASの活性化が確認される黒色腫においては,mTOR複合体1の恒常的な活性化によりオートファジーが抑制されているらしい.したがって,黒色腫はアミノ酸プールの減少に応答したオートファジーを誘導できず細胞死をひき起こす81).種々のがんにおけるオートファジーの生理的な役割は,環境やステージ,そして,遺伝学的な背景により異なるのだろう.事実,最近,p53の欠失の有無により膵がんにおけるオートファジーの役割が異なることが明らかになった82).
おわりに
オートファジー関連遺伝子の同定をはじめ,オートファジー研究分野における日本人の貢献は大きい.しかし現在,欧米およびアジアの各国ではオートファジーの研究において基礎レベルのみならずトランスレーショナルリサーチも活発に行われており,臨床への応用に関してわが国は後塵を拝している感がある.とくに,がん細胞においてはオートファジーによるアミノ酸の供給やミトコンドリアなどのオルガネラの品質管理がその増殖に決定的であり,既存の抗がん剤とオートファジー阻害剤(正確には,リソソーム酸性化阻害剤)をあわせたがん治療の臨床試験がすでに欧米において開始されている.また,世界中でオートファジーに特異的な活性化剤および阻害剤のスクリーニングが行われ,いくつかは動物レベルでその効果が確認されている.しかし,オートファジーの臨床への応用にはいくつかの問題点が残っている.オートファジーの分子機構には未解明の謎がいまだ山積しており,その阻害あるいは亢進によりどのような副作用があるのかも不透明である.現在まで,オートファジーの活性をモニターできるマーカーもプローブも存在せず,薬効の判定ができない.遺伝子改変動物の解析によりおのおのの組織におけるオートファジーの生理機能の大筋は判明しつつあるが,ヒトの病態におけるその意義はほとんど手つかずの状態であり,さらなる解析が必要であろう.
文 献
- Mizushima, N., Yoshimori, T. & Ohsumi, Y.: The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol., 27, 107-132 (2011)[PubMed]
- Dennis, M. D., Baum, J. I., Kimball, S. R. et al.: Mechanisms involved in the coordinate regulation of mTORC1 by insulin and amino acids. J. Biol. Chem., 286, 8287-8296 (2011)[PubMed]
- Diaz-Troya, S., Perez-Perez, M. E., Florencio, F. J. et al.: The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy, 4, 851-865 (2008)[PubMed]
- Noda, T. & Ohsumi, Y.: Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem., 273, 3963-3966 (1998)[PubMed]
- Inoki, K. & Guan, K. L.: Complexity of the TOR signaling network. Trends Cell Biol., 16, 206-212 (2006)[PubMed]
- Inoki, K., Li, Y., Zhu, T. et al.: TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol., 4, 648-657 (2002)[PubMed]
- Long, X., Lin, Y., Ortiz-Vega, S. et al.: Rheb binds and regulates the mTOR kinase. Curr. Biol., 15, 702-713 (2005)[PubMed]
- Zhang, Y., Gao, X., Saucedo, L. J. et al.: Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol., 5, 578-581 (2003)[PubMed]
- Kim, E., Goraksha-Hicks, P., Li, L. et al.: Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol., 10, 935-945 (2008)[PubMed]
- Sancak, Y., Bar-Peled, L., Zoncu, R. et al.: Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell, 141, 290-303 (2010)[PubMed]
- Sancak, Y., Peterson, T. R., Shaul, Y. D. et al.: The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science, 320, 1496-1501 (2008)[PubMed]
- Bonfils, G., Jaquenoud, M., Bontron, S. et al.: Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol. Cell, 46, 105-110 (2012)[PubMed]
- Duran, R. V., Oppliger, W., Robitaille, A. M. et al.: Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell, 47, 349-358 (2012)[PubMed]
- Han, J. M., Jeong, S. J., Park, M. C. et al.: Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell, 149, 410-424 (2012)[PubMed]
- Zoncu, R., Bar-Peled, L., Efeyan, A. et al.: mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science, 334, 678-683 (2011)[PubMed]
- Bar-Peled, L., Schweitzer, L. D., Zoncu, R. et al.: Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell, 150, 1196-1208 (2012)[PubMed]
- Hosokawa, N., Hara, T., Kaizuka, T. et al.: Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell, 20, 1981-1991 (2009)[PubMed]
- Naito, T., Kuma, A. & Mizushima, N.: Differential contribution of insulin and amino acids to the mTORC1-autophagy pathway in the liver and muscle. J. Biol. Chem., 288, 21074-21081 (2013)[PubMed]
- Gwinn, D. M., Shackelford, D. B., Egan, D. F. et al.: AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell, 30, 214-226 (2008)[PubMed]
- Inoki, K., Zhu, T. & Guan, K. L.: TSC2 mediates cellular energy response to control cell growth and survival. Cell, 115, 577-590 (2003)[PubMed]
- Kim, J., Kim, Y. C., Fang, C. et al.: Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell, 152, 290-303 (2013)[PubMed]
- Egan, D. F., Shackelford, D. B., Mihaylova, M. M. et al.: Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science, 331, 456-461 (2011)[PubMed]
- Kuma, A., Hatano, M., Matsui, M. et al.: The role of autophagy during the early neonatal starvation period. Nature, 432, 1032-1036 (2004)[PubMed]
- Ezaki, J., Matsumoto, N., Takeda-Ezaki, M. et al.: Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy, 7, 727-736 (2011)[PubMed]
- Guo, J. Y., Chen, H. Y., Mathew, R. et al.: Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev., 25, 460-470 (2011)[PubMed]
- Tsukamoto, S., Kuma, A., Murakami, M. et al.: Autophagy is essential for preimplantation development of mouse embryos. Science, 321, 117-120 (2008)[PubMed]
- Baba, M., Osumi, M., Scott, S. V. et al.: Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J. Cell Biol., 139, 1687-1695 (1997)[PubMed]
- Sakai, Y., Koller, A., Rangell, L. K. et al.: Peroxisome degradation by microautophagy in Pichia pastoris: identification of specific steps and morphological intermediates. J. Cell Biol., 141, 625-636 (1998)[PubMed]
- Nakagawa, I., Amano, A., Mizushima, N. et al.: Autophagy defends cells against invading group A Streptococcus. Science, 306, 1037-1040 (2004)[PubMed]
- Bjorkoy, G., Lamark, T., Brech, A. et al.: p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol., 171, 603-614 (2005)[PubMed]
- Komatsu, M., Kurokawa, H., Waguri, S. et al.: The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol., 12, 213-223 (2010)[PubMed]
- Okamoto, K., Kondo-Okamoto, N. & Ohsumi, Y.: Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell, 17, 87-97 (2009)[PubMed]
- Narendra, D., Tanaka, A., Suen, D. F. et al.: Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol., 183, 795-803 (2008)[PubMed]
- Hampe, J., Franke, A., Rosenstiel, P. et al.: A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet., 39, 207-211 (2007)[PubMed]
- Rioux, J. D., Xavier, R. J., Taylor, K. D. et al.: Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet., 39, 596-604 (2007)[PubMed]
- Hanada, T., Noda, N. N., Satomi, Y. et al.: The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem., 282, 37298-37302 (2007)[PubMed]
- Fujita, N., Saitoh, T., Kageyama, S. et al.: Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: analysis of the organization of the Atg16L1 complex in fibroblasts. J. Biol. Chem., 284, 32602-32609 (2009)[PubMed]
- Mizushima, N., Noda, T. & Ohsumi, Y.: Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J., 18, 3888-3896 (1999)[PubMed]
- Murthy, A., Li, Y., Peng, I. et al.: A Crohn's disease variant in Atg16l1 enhances its degradation by caspase 3. Nature, 506, 456-462 (2014)[PubMed]
- Adolph, T. E., Tomczak, M. F., Niederreiter, L. et al.: Paneth cells as a site of origin for intestinal inflammation. Nature, 503, 272-276 (2013)[PubMed]
- Cadwell, K., Liu, J. Y., Brown, S. L. et al.: A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature, 456, 259-263 (2008)[PubMed]
- Saitoh, T., Fujita, N., Jang, M. H. et al.: Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature, 456, 264-268 (2008)[PubMed]
- Geisler, S., Holmstrom, K. M., Skujat, D. et al.: PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol., 12, 119-131 (2010)[PubMed]
- Kawajiri, S., Saiki, S., Sato, S. et al.: PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett., 584, 1073-1079 (2010)[PubMed]
- Matsuda, N., Sato, S., Shiba, K. et al.: PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol., 189, 211-221 (2010)[PubMed]
- Narendra, D. P., Jin, S. M., Tanaka, A. et al.: PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol., 8, e1000298 (2010)[PubMed]
- Vives-Bauza, C., Zhou, C., Huang, Y. et al.: PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA, 107, 378-383 (2010)[PubMed]
- Greene, A. W., Grenier, K., Aguileta, M. A. et al.: Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep., 13, 378-385 (2012)[PubMed]
- Jin, S. M., Lazarou, M., Wang, C. et al.: Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol., 191, 933-942 (2010)[PubMed]
- Okatsu, K., Oka, T., Iguchi, M. et al.: PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun., 3, 1016 (2012)[PubMed]
- Koyano, F., Okatsu, K., Kosako, H. et al.: Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature, 510, 162-166 (2014)[PubMed] [新着論文レビュー]
- Yoshii, S. R., Kishi, C., Ishihara, N. et al.: Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem., 286, 19630-19640 (2011)[PubMed]
- Haack, T. B., Hogarth, P., Kruer, M. C. et al.: Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet., 91, 1144-1149 (2012)[PubMed]
- Saitsu, H., Nishimura, T., Muramatsu, K. et al.: De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet., 45, 445-449 (2013)[PubMed] [新着論文レビュー]
- Guan, J., Stromhaug, P. E., Georg,e M. D. et al.: Cvt18/Gsa12 is required for cytoplasm-to-vacuole transport, pexophagy, and autophagy in Saccharomyces cerevisiae and Pichia pastoris. Mol. Biol. Cell, 12, 3821-3838 (2001)[PubMed]
- Proikas-Cezanne, T., Waddell, S,, Gaugel, A. et al.: WIPI-1α (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 23, 9314-9325 (2004)[PubMed]
- Dove, S. K., Piper, R. C., McEwen, R. K. et al.: Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. EMBO J., 23, 1922-1933 (2004)[PubMed]
- Polson, H. E., de Lartigue, J., Rigden, D. J. et al.: Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy, 6, 506-522 (2010)[PubMed]
- Baskaran, S., Ragusa, M. J., Boura, E et al.: Two-site recognition of phosphatidylinositol 3-phosphate by PROPPINs in autophagy. Mol. Cell, 47, 339-348 (2012)[PubMed]
- Krick, R., Busse, R. A., Scacioc, A. et al.: Structural and functional characterization of the two phosphoinositide binding sites of PROPPINs, a β-propeller protein family. Proc. Natl. Acad. Sci. USA, 109, E2042- E2049 (2012)[PubMed]
- Watanabe, Y., Kobayashi, T., Yamamoto, H. et al.: Structure-based analyses reveal distinct binding sites for Atg2 and phosphoinositides in Atg18. J. Biol. Chem., 287, 31681-31690 (2012)[PubMed]
- Hayflick, S. J., Kruer, M. C., Gregory, A. et al.: β-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain, 136, 1708-1717 (2013)[PubMed]
- Cullup, T., Kho, A. L., Dionisi-Vici, C. et al.: Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat. Genet., 45, 83-87 (2013)[PubMed]
- Tian, Y., Li, Z., Hu, W. et al.: C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell, 141, 1042-1055 (2010)[PubMed]
- Zhao, H., Zhao, Y. G., Wang, X. et al.: Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J. Cell Biol., 200, 731-741 (2013)[PubMed]
- Liang, X. H., Jackson, S., Seaman, M. et al.: Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature, 402, 672-676 (1999)[PubMed]
- Inami, Y., Waguri, S., Sakamoto, A. et al.: Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol., 193, 275-284 (2011)[PubMed]
- Takamura, A., Komatsu, M., Hara, T. et al.: Autophagy-deficient mice develop multiple liver tumors. Genes Dev., 25, 795-800 (2011)[PubMed]
- Li, L., Shen, C., Nakamura, E. et al.: SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell, 24, 738-750 (2013)[PubMed]
- Moscat, J. & Diaz-Meco, M. T.: p62 at the crossroads of autophagy, apoptosis, and cancer. Cell, 137, 1001-1004 (2009)[PubMed]
- Duran, A., Linares, J. F., Galvez, A. S. et al.: The signaling adaptor p62 is an important NF-κB mediator in tumorigenesis. Cancer Cell, 13, 343-354 (2008)[PubMed]
- Ling, J., Kang, Y., Zhao, R. et al.: KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell, 21, 105-120 (2012)[PubMed]
- Ichimura, Y., Waguri, S., Sou, Y. S. et al.: Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell, 51, 618-631 (2013)[PubMed] [新着論文レビュー]
- Mitsuishi, Y., Taguchi, K,, Kawatanim Y. et al.: Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell, 22, 66-79 (2012)[PubMed] [新着論文レビュー]
- Kimmelman, A. C.: The dynamic nature of autophagy in cancer. Genes Dev., 25, 1999-2010 (2011)[PubMed]
- Yang, S., Wang, X., Contino, G. et al.: Pancreatic cancers require autophagy for tumor growth. Genes Dev., 25, 717-729 (2011)[PubMed]
- Amaravadi, R. K., Yu, D., Lum, J. J. et al.: Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Invest., 117, 326-336 (2007)[PubMed]
- Wei, H., Wei, S., Gan, B. et al.: Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev., 25, 1510-1527 (2011)[PubMed]
- Elgendy, M., Sheridan, C., Brumatti, G. et al.: Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell, 42, 23-35 (2011)[PubMed]
- Young, A. R., Narita, M., Ferreira, M. et al.: Autophagy mediates the mitotic senescence transition. Genes Dev., 23, 798-803 (2009)[PubMed]
- Sheen, J. H., Zoncu, R., Kim, D. et al.: Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell, 19, 613-628 (2011)[PubMed]
- Rosenfeldt, M. T., O'Prey, J., Morton, J. P. et al.: p53 status determines the role of autophagy in pancreatic tumour development. Nature, 504, 296-300 (2013)[PubMed]
著者プロフィール
略歴:2011年 大阪大学大学院生命機能研究科 修了,同年 東京都医学総合研究所 研究員を経て,2014年より新潟大学大学院医歯学総合研究科 助教.
研究テーマ:生体におけるオートファジーの生理的な意義および分子機構.とくに,選択的オートファジーに着目し,疾患とのかかわりを明らかにしていきたい.
小松 雅明(Masaaki Komatsu)
新潟大学大学院医歯学総合研究科 教授.
研究室URL:http://www.med.niigata-u.ac.jp/bc1/welcome.html
© 2014 蔭山 俊・小松雅明 Licensed under CC 表示 2.1 日本
