ゲノム編集がひらく遺伝子改変マウスの未来
2014/07/15
伊川 正人
(大阪大学微生物病研究所 附属感染動物実験施設)
email:伊川正人
領域融合レビュー, 3, e008 (2014) DOI: 10.7875/leading.author.3.e008
Masahito Ikawa: Genome editing in mice opens a new era for biological and biomedical researches.

遺伝子改変動物を用いた研究の醍醐味は,培養細胞や試験管内ではみられないダイナミックな高次生命現象を,個体レベルで観察あるいは解析できることにある.とくに,ヒトと同じ哺乳類に属するマウスは,全ゲノム塩基配列が明らかにされているだけでなく,ES細胞における相同組換えを利用した標的遺伝子の破壊が可能であることから,遺伝子機能解析ツールとしてだけでなく,ヒトの疾患の病態モデルとして,生命科学研究および医学研究の礎を担っている.さらに最近では,黒船襲来ともいうべきゲノム編集技術,とくにRNAにより標的配列を認識するCRISPR/Cas法の開発により,1塩基レベルから染色体レベルでの遺伝子の挿入,欠損,置換,さらには,エフェクターとの組合せによる転写制御,ゲノムの可視化,エピゲノム修飾の操作などの可能性が大きく広がっている.このレビューでは,従来の遺伝子改変マウスの作製技術をふまえ,ゲノム編集がひらく遺伝子改変マウスの未来について解説する.
マウスは約20~30 gと小さく穏やかな性質をもち多産な動物である.また,性成熟(雄で約6週間,雌で約4週間)や妊娠期間(約20日間)を含めて比較的ライフサイクルが短いことから,古くから実験動物として供されてきた.さらに,全ゲノム塩基配列の解読によりヒトの遺伝子の99%はマウスにも保存されていることが明らかにされたこと,体外受精や胚操作などの生殖工学的な手法も確立されていることなどもあり,マウスは遺伝学的にも微生物学的にも制御されたすばらしい実験動物であるといえる.
個体レベルにおいてマウスのゲノム遺伝子を操作する試みは古く,さまざまな手法が活用されてきた.もっとも古典的な方法は自然変異マウスのスクリーニングであり,胸腺を欠損するヌードマウスや,T細胞およびB細胞を欠損するSCIDマウスなどが有名である.つづいて,放射線の照射や化学変異原の投与などの人為突然変異法が開発された.たとえば,エチルニトロソ尿素(ethylnitrosourea:ENU)の投与は自然変異の約100倍~1000倍の確率で点変異を誘発し,ヌル欠損変異だけでなくアミノ酸置換変異なども得られる.方法は簡単で,エチルニトロソ尿素を投与した雄と野生型の雌を交配させて得られる子孫を表現型によりスクリーニングする.しかしながら,ゲノムにおいて変異を同定することは非常に困難であった.近年の塩基配列解析技術の飛躍的な進展にともない,ランダムに導入された変異をさきにスクリーニングするというアプローチも可能にはなっているが1),思いどおりの変異を導入できないという難点は残る.なお,自然変異マウスおよび人為突然変異マウスはいずれも遺伝子組換え実験の規制にとらわれず,野生型のマウスと同様に扱うことができる.
変異マウスを用いて表現型からその原因となる遺伝子を探りあてる順遺伝学(フォワードジェネティクス)に対し,遺伝子を改変して表現型との関連を探るアプローチを逆遺伝学(リバースジェネティクス)という.1970年代に外来の遺伝子を導入したトランスジェニックマウスが,1980年台後半に内在性の遺伝子を破壊したノックアウトマウスが開発されたことにより,逆遺伝学は大きく進展した.さらに2010年台に入り,ゲノム編集技術を活用した遺伝子改変マウスの可能性が大きく広がっている.以下,遺伝子組換えマウスの作製技術とその可能性について解説する.
一般的には,トランス遺伝子とよばれる外来性の遺伝子を宿主のゲノムに人為的に組み込み,これが世代をこえて受け継がれるマウスをさす.宿主がもたないタンパク質を発現させたり,野生型あるいは変異型のタンパク質を過剰に発現させたりする目的で作製されることが多い.たとえば,筆者らが開発した蛍光タンパク質EGFPを発現するグリーンマウスは移植やがん転移など幅広い基礎研究に,また,rasH2マウスは発がん試験に用いられている.組織特異的なプロモーターや薬剤誘導型のプロモーターを使うことにより,より複雑な遺伝子発現の制御も可能となる.
トランスジェニックマウスは受精卵の前核にガラス管を用いて直鎖状のDNAを直接注入することにより作製することが多い(図1a).トランス遺伝子はゲノムの1箇所にタンデムに複数のコピーが組み込まれることが多く,挿入位置やコピー数により発現パターンや強度が左右される.数は少ないが,レンチウイルスベクターを受精卵に感染させる手法も使われる(図1b).この場合は,ゲノムの複数の個所に1コピーずつ組み込まれるため,位置効果を相殺することができる.また,前核に注入する方法に比べ作製効率が10倍近く改善されることから,稀少な変異マウス系統を使うときや第1世代での発現解析などに威力を発揮する.
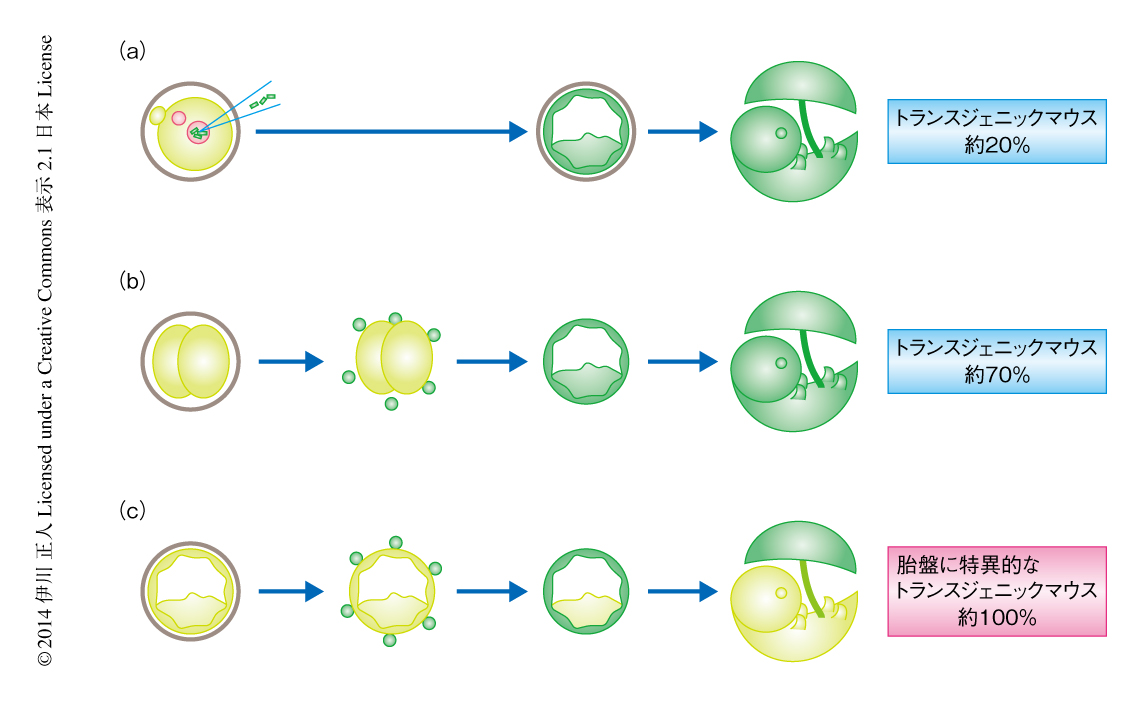
なお,従来の方法では胎仔と胎盤の両方に遺伝子が導入されるため,胚性致死などの解析が困難であった.筆者らは,発生の進んだ胚盤胞期の胚にレンチウイルスベクターを感染させれば,胎仔に遺伝子導入することなく胎盤に特異的に遺伝子導入することができることを見い出した(図1c).妊娠期の遺伝子機能解析ツールとして活用が期待される.
ノックアウトマウスは内在性の遺伝子が破壊されたマウスの総称として使われており,点変異によるフレームシフト,ナンセンス変異,ミスセンス変異による機能の欠失,のちに述べるゲノム編集技術などにより遺伝子を破壊されたマウスを含む.一般的には,内在性の遺伝子を破壊したES細胞(embryonic stem cell,胚性幹細胞)からキメラマウスを介して生殖系列に変異した遺伝子を伝達するマウスをさすことが多い.
1981年,マウスの胚盤胞からES細胞が樹立され,胚盤胞に混合すればキメラマウスが作製できること,さらに,交配によりES細胞に由来する子孫を得られることが報告された.そののち,薬剤により選択することのできる培養細胞株の利点を生かし,ES細胞を用いたマウスの遺伝子破壊法として有名な2つの方法が開発された.ランダムに遺伝子を破壊する遺伝子トラップ法と,標的とした遺伝子を破壊する遺伝子ターゲティング法である.遺伝子トラップ法は,外来性の薬剤耐性遺伝子がゲノムにランダムに挿入される際に,偶然に内在性の遺伝子座に挿入されたものを薬剤により選択する.遺伝子ターゲティング法は,外来性の薬剤耐性遺伝子の両側にゲノムと相同性をもつ領域を付加し,相同組換えにより内在性の遺伝子を薬剤耐性遺伝子と置き換える方法であり,目的とする遺伝子の機能を個体レベルで解析する逆遺伝学の典型例とされる.1980年台の終わりから1990年代にかけて,ノックアウトマウスに関する多くの論文がトップジャーナルに掲載され,個体レベルでの遺伝子の機能解析にはノックアウトマウスが常道とされるようになった.2007年には,ノックアウトマウスの開発者であるMartin Evans,Oliver Smithies,Mario Capecchiにノーベル医学生理学賞が授与されている.
遺伝子トラップ法においては,当初は薬剤耐性遺伝子とポリA付加シグナルをつないだカセットを用いて内在性のプロモーターをとらえるプロモータートラップ法が主流であったが,ES細胞で発現している遺伝子しか破壊できないという欠点があった(図2a).そこで考案されたのが,普遍的に発現するプロモーターの後ろにレポーター遺伝子を挿入したカセットを用いてポリAをとらえるポリAトラップ法で,原理的にはすべての遺伝子をトラップして破壊できる.しかし,ポリAトラップ法では薬剤耐性遺伝子のもつ終止コドンの後ろに内在性のエキソンが連なるため,ナンセンス変異依存性mRNA分解機構(non-sense mediated mRNA decay:NMD)によりmRNAの不安定化をひき起こすという問題があった.現在では,薬剤耐性遺伝子の後ろにリボソームエントリー部位と3フレームのATGを付加することなどで問題は解決されている2).遺伝子トラップカセットの導入には,直線化プラスミドのエレクトロポレーション法も利用されるが,細胞の生存率や導入効率からレトロウイルスベクターを使った方法が好まれる.最近は,Sleeping BeautyやPiggyBacなどのトランスポゾン系を使ってゲノムに組み込むアプローチも開発されており,その場合は,表現型を観察したのちにトラップベクターを抜き取って機能の回復を確認できるという利点がある(図2a).
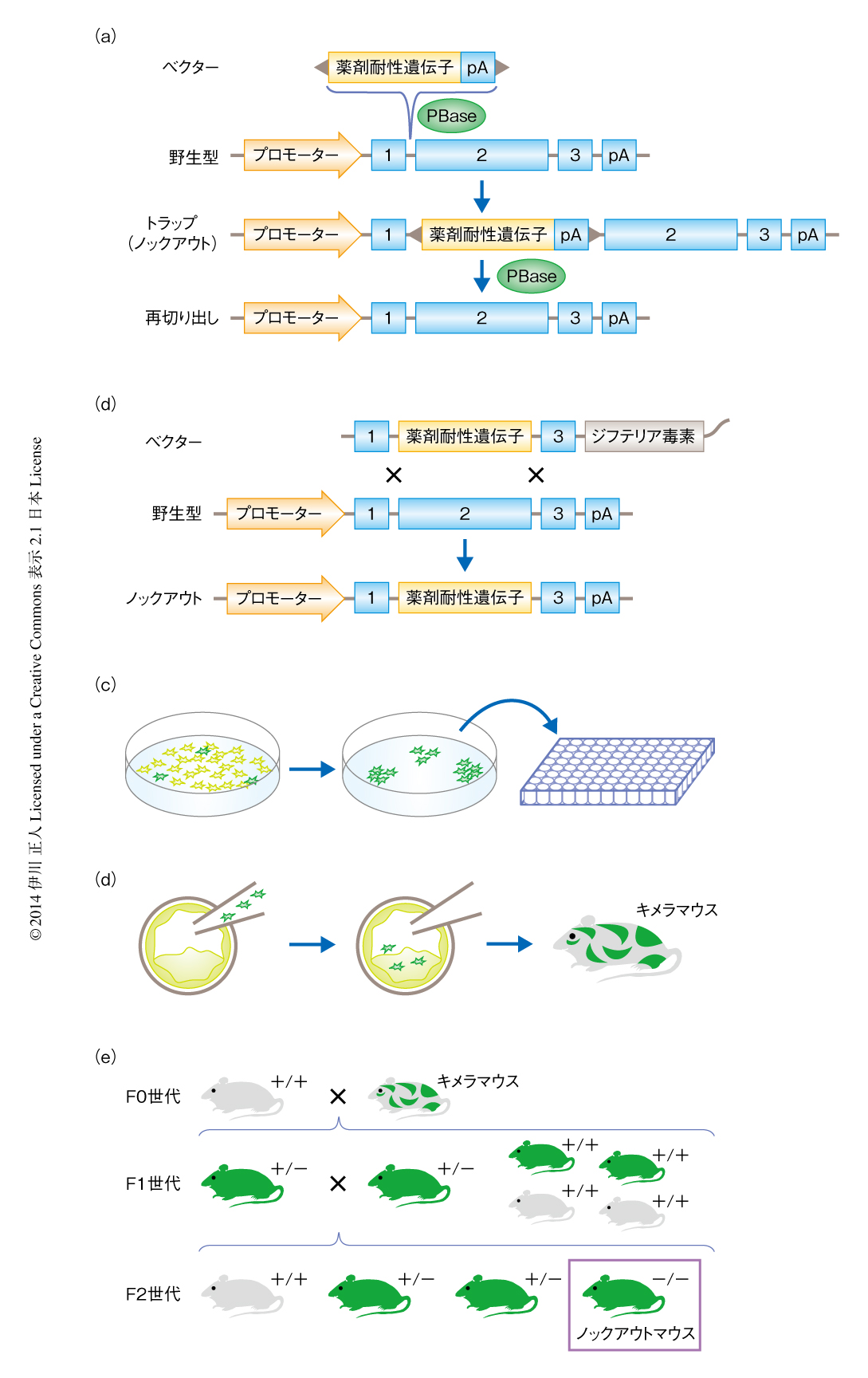
遺伝子トラップ法は同じベクターをくり返しES細胞に導入することによりランダムに遺伝子を破壊するのに対し,遺伝子ターゲティング法では標的とする遺伝子ごとにデザインされたベクターを作製してES細胞へ導入し,薬剤耐性遺伝子および薬剤感受性遺伝子(あるいは,自殺遺伝子)を組み合わせた正の選択および負の選択により相同組換えを起こしたES細胞を濃縮する(図2b).つぎに,遺伝子を導入したES細胞を薬剤により選択しクローン化する(図2c).クローン化したES細胞からゲノムDNAを抽出してPCR法やサザンブロット法により相同組換えを確認する.ES細胞を初期胚と凝集し,偽妊娠マウスに移植してキメラマウスを作製する(図2d).当初,汎用された129系統のES細胞はC57BL/6系統の胚盤胞期の胚に注入してキメラマウスを作製していたが,C57BL/6系統のES細胞は,ICR系統の8細胞期の胚と凝集させるほうが生殖系列に寄与しやすいようである.得られたキメラマウスを野生型マウスと交配させて,標的遺伝子に変異をもつF1マウスを得たのち,ヘテロF1マウスの雌雄を交配してホモ変異をもつF2マウスを得る(図2e).ベクターの構築から,ES細胞における相同組換え,キメラマウスの作製と交配による生殖系列への寄与,さらに,交配によるホモ欠損マウスを得るためには,年単位の実験になることも多い.
マウスではES細胞の培養技術,遺伝子改変技術,キメラ動物の作製技術が確立しているが,ほかの動物種ではそうともかぎらない.たとえばラットでは,マウスから遅れること15年,2003年になってようやくES細胞を使った遺伝子ノックアウトが報告された.ES細胞以外の細胞を用いた遺伝子ターゲティング法も開発されている.iPS細胞を用いればES細胞と同様の実験ができることはいうまでもないが,GS細胞(germ line stem cell,雄性生殖幹細胞)において標的遺伝子を破壊し,それを精巣に移植して精子を形成させ次世代を得ることもできる3).なお,家畜動物などでは,遺伝子ターゲティングした胎仔繊維芽細胞からクローン技術により個体を復元する方法も一般的である.
遺伝子ノックアウト実験の醍醐味は,表現型をみるまで標的遺伝子の重要性がわからないことであろう.筆者らもこれまで数多くのノックアウトマウスを作製してきたが,in vitroにおける実験の結果から誰もが必須と信じて疑わなかった遺伝子が不要であったり,まったく予期せぬ結果から生命現象に必須の遺伝子がみつかったりしたケースも少なくない.ノックアウトマウスの結果しだいでは定説を一発逆転できることから,予想どおりの表現型より,むしろ予想外の結果が得られたほうが大きな発見につながる.
遺伝子ターゲティングの際の重要なポイントは,生殖系列に寄与できる未分化な状態を維持したES細胞の培養と,効率的な相同組換えである.ES細胞の培養については,由来するマウスの系統や樹立されたES細胞株などによって異なり,培地や血清のロットにも左右される.そのため,遺伝子操作と継代の過程において性質が変化してキメラマウスが得られなかったり,キメラマウスが誕生しても遺伝子改変したES細胞に由来する仔が得られず,実験が振り出しにもどるケースも少なくなかった.効率的な相同組換えについては,組換えの効率を上げる目的で相同領域を長くとるため(両側合計で7~10 kbp),全長が20 kbp近いプラスミドの切り貼りとなり,ベクターの構築と配列の確認に手間と時間を要するという問題点があった.また,相同組換えの頻度は標的遺伝子座やベクターのもつ相同領域の配列などによっても大きく異なるため,トラブルシューティングは経験や勘にたよる部分も多く,運が悪いのか腕が悪いのかわからないなど初心者にはむずかしい実験であった.
さらに残念なことに,コストや労力をかけて作製したがゆえに,得られたノックアウトマウスを研究材料として共有しない研究室もあり,有名な遺伝子のノックアウトマウスは多くの研究室で重複して作製された.また,表現型のみられないノックアウトマウスの情報は表にでないため,同じノックアウトマウスが異なる研究室でくり返し作製されるといったむだも生じていた.
これらの背景から,少ない拠点においてすべての遺伝子を網羅的にノックアウトして人類共通の財産にしようという国際ノックアウトマウスプロジェクトが始動した4).米国やヨーロッパを中心に活動が広がり,2006年ごろ,IKMC(International Knockout Mouse Consortium)が国際プロジェクトとして活動を本格化した.当初は遺伝子トラップ法が主流であったが,相同組換え率の高いベクターの開発や,ベクター構築の効率化あるいは機械化により,遺伝子ターゲティング法も採用されている.IKMCで採用された遺伝子ターゲティングベクターの多くが組織特異的あるいは時期特異的に遺伝子を破壊できるコンディショナルノックアウトタイプであり,内在性のプロモーターによりレポーターとしてLacZ遺伝子を発現するといったくふうもなされている.
なお,2011年から国際ノックアウトマウスプロジェクトはIMPC(International Mouse Phenotyping Consortium,URL:http://www.mousephenotype.org/)にひき継がれ,これまでに,IKMCとあわせて2万近い遺伝子について組換えES細胞株が樹立されている.今後は,キメラマウスを介したノックアウトマウスの作製と1次的な表現型の解析が主体となり,解析の結果は世界中に公開される.当初5年で4000系統の解析を終えるという目標が設定されているが,細胞レベルとは異なり個体レベルの実験には莫大な費用と時間がかかる.2014年6月時点では約300系統の表現型の公開にとどまっており,今後の進展が期待される.
欧米各国が莫大な予算を投じて国際ノックアウトマウスプロジェクトを推進するかたわら,新たな遺伝子改変技術が産声をあげていた.人工制限酵素の開発と応用による標的遺伝子の改変技術,つまり,ゲノム編集技術である.原理的には,ゲノムの2本鎖DNAを切断し,その修復機構のエラーを利用して遺伝子を効率的に改変する(図3a).非相同末端結合(nonhomologous end-joining:NHEJ)経路による修復では,切断をうけたDNA末端を直接つなぎ合わせる際に,数塩基の欠失あるいは挿入などのエラーが起こりやすい.また,相同組換え(homologous recombination:HR)経路による修復ではエラーは少ないが,相同性をもつ1本鎖DNAあるいは2本鎖DNAを人為的に導入しておくとあやまってゲノムに取り込まれることがある.ゲノム編集技術では,このような2本鎖DNA切断とその修復のエラーをうまく利用して遺伝子改変を行う5).
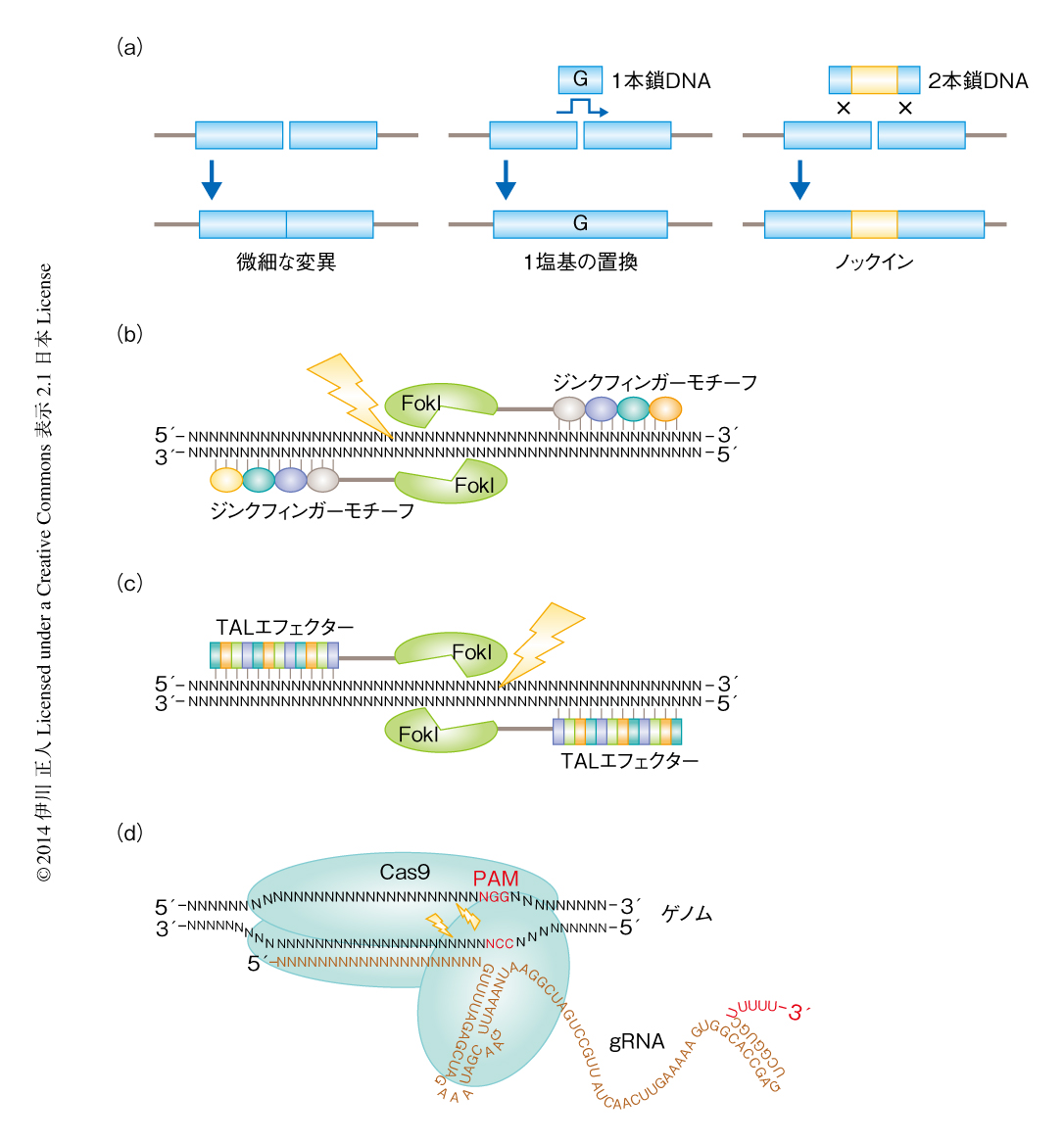
ゲノム編集といえばZFN(zinc finger nuclease)が最初と思う人が多いかもしれないが,それ以前から,2本鎖DNA切断を利用した遺伝子改変は試みられていた.1996年には,18塩基を特異的に認識するI-SceIメガヌクレアーゼを用いた2本鎖DNA切断により相同組換えの効率の改善することが報告されている.しかし,メガヌクレアーゼは標的配列をデザインすることができないため,認識配列をさきにゲノムに組み込む必要があるなど,実用的にはほとんど意味のないものであった.同じく1996年に開発されたのがZFNである.ジンクフィンガーモチーフとよばれるDNA結合ドメインとFokIヌクレアーゼで構成された人工キメラタンパク質であり,標的遺伝子を認識し切断する(図3b).しかし,ZFNではDNA結合ドメインがデザインどおりにDNAを認識しないことが多く,ベクターの構築がむずかしいとの欠点があった.また,市販品が非常に高額であったため,一般の研究者には高嶺の花であることも多かった.
つぎに登場したのが,ZFNのDNA結合ドメインを植物病原細菌であるXanthomonas属細菌のもつTALEエフェクターに代えたTALENである(図3c).核酸のA,C,G,Tそれぞれ1塩基に特異的に対応するユニットを標的配列にあわせて組み合わせるだけで構築できることから,一般の研究者にも利用できるレベルになった.さらに数塩基を認識するモジュールを組み合わせる改善もなされている.また,34アミノ酸残基からなるユニットの4番目と32番目のアミノ酸残基のバリエーションに注意してモジュールを構築することにより,活性の高いTALENを効率よく作製できるPlatinum TALENも開発されている6)(URL:http://www.addgene.org/TALEN/PlatinumGate/).
ZFNやTALENといった人工制限酵素は,受精卵に注入するだけで内在性の遺伝子を破壊できることから,ホヤ,カエル,ゼブラフィッシュなど,多くのモデル生物において利用されている.詳細については末尾にあげた参考図書を参照されたい.
ZFNやTALENの登場により“人工制限酵素”や“ゲノム編集”という言葉が研究者の耳目をあつめ,また,画期的な遺伝子改変技術としての評価から,2011年にはNature Method誌のMethod of the Year,2012年にはScience誌のBreakthrough of the yearにとりあげられた.そのような状況のなか,次世代のゲノム編集技術として開発されたのがCRISPR/Cas法である.そもそも,CRISPR/Cas系は古細菌などがもつ獲得免疫機構であり,ファージなどを介して侵入する外来性のDNAを断片化して自らのゲノムに組み込み,2度目の感染のときに,この外来性のDNAの断片から発現する2つの小分子RNAを用いて標的DNAを認識し,Cas9ヌクレアーゼをリクルートしてこれを切断する.2013年2月には,2つの小分子RNAをひとつのキメラRNAとし(guide RNA:gRNA,あるいは,single guide RNA:sgRNAとよばれる),Cas9との2つの因子だけでゲノムDNAの切断活性を発揮できることが示された7,8)(図3d).
ZFNやTALENはDNA結合ドメインと切断ドメインを融合タンパク質として機能させるため標的遺伝子ごとに複雑なベクターの構築が必要なのに対し,CRISPR/Cas法では標的配列に対応するgRNAの5’末端の20塩基を変更するだけで標的遺伝子を変えることができる.この手軽さと低コスト,高い切断効率と再現性から,CRISPR/Cas法は瞬く間に生命科学研究者に広まった.筆者らはプラスミドpX330(URL:http://www.addgene.org/42230/)を用いているが,pX330にはgRNA発現カセットおよびコドンをヒト化したCas9発現カセットが搭載されており,合成オリゴDNAを導入するだけでノックアウトベクターを構築できる(図4a).
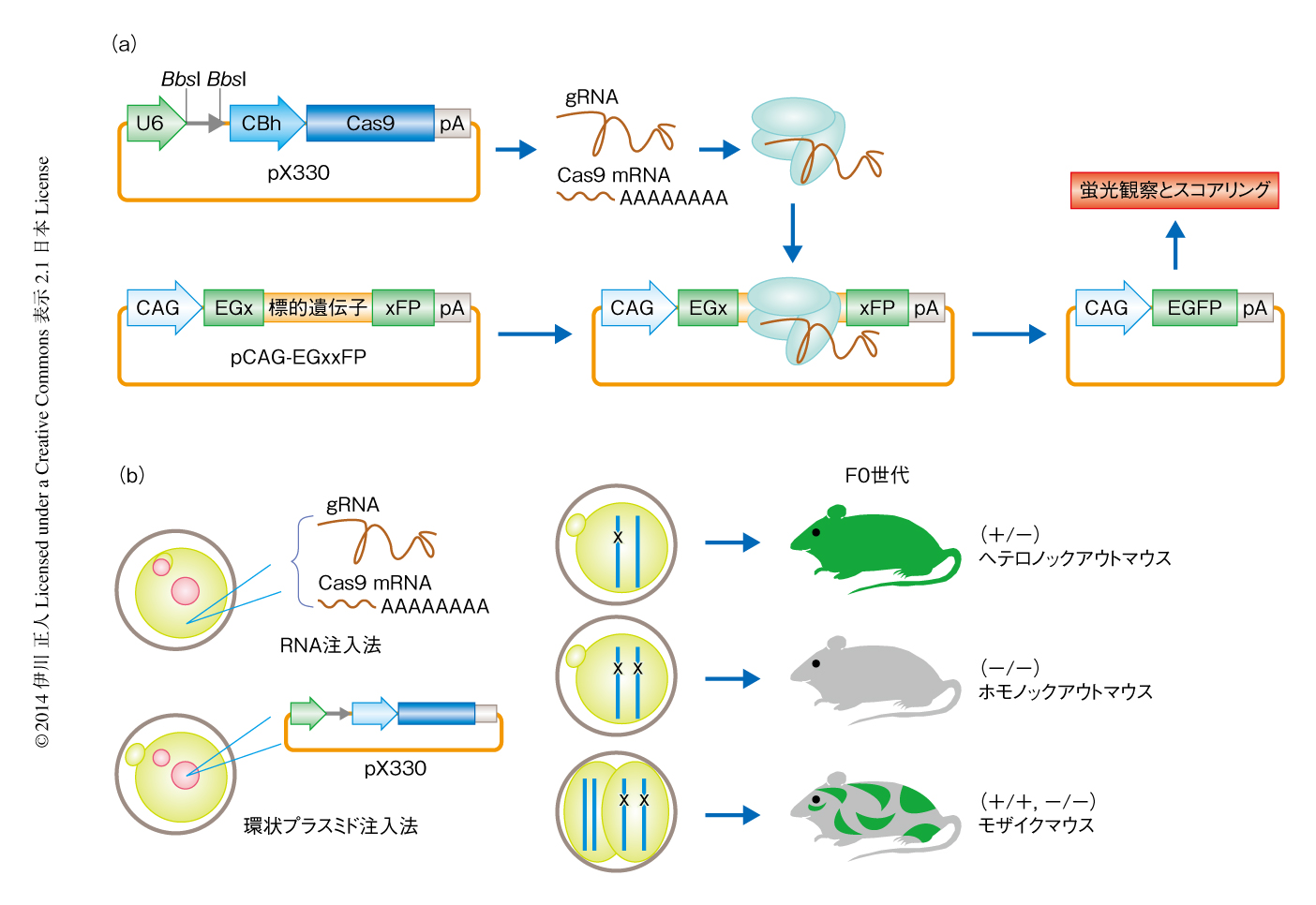
CRISPR/Cas法が哺乳類培養細胞において機能することが示されてから半年も経たない2013年5月,Cas9をコードするmRNAとgRNAを受精卵の細胞質に注入することによりノックアウトマウスが得られるという論文が報告された9)(図4b).当時,筆者らも,RNA合成のステップをスキップしてpX330プラスミドを受精卵の前核にマイクロインジェクションする簡易法を独自に開発していたが,2番手の論文ということで予想以上に審査が厳しく,論文発表までに半年を要した10).CRISPR/Cas法を用いたノックアウトマウス作製のメリットはその簡便性とスピードであり,実際に,合成オリゴDNAの発注からpX330ベクターの構築,培養細胞における切断活性の検討に約1週間,それに受精卵への注入から出産までの3週間をくわえた約1カ月でノックアウトマウスが作製できる.さらに,変異マウスの約半数において両方の対立遺伝子に変異が導入されている点は特筆すべきであり,ES細胞を使うと交配だけでも半年を要することを考えると,ファウンダー世代でホモ欠損マウスが得られるのは夢のようである.筆者らの経験では,100個の受精卵を処理すれば約10匹が生まれ,5匹程度が変異マウス,うち2匹程度が両方の対立遺伝子に変異をもつ10,11).なお,受精卵ではなく,ES細胞へのプラスミドのトランスフェクションでは,pX330が一過性に導入されたES細胞の約8割で両方の対立遺伝子に変異が認められる.
CRISPR/Cas法の注意点としては,標的とする配列に近似した配列(オフターゲット)を切断してしまうリスクがある.ZFNやTALENでは,センス鎖とアンチセンス鎖を認識する2つのユニットが同時に認識してはじめてFokIによる切断が起こるのに対し,片側のDNA鎖のみを認識するCRISPR/Cas法の特異性は低いとされ,がん細胞などの培養細胞などではオフターゲットの切断が多く報告されている.しかし,受精卵やES細胞などではDNA修復能力が高いのか,それほど問題にならないようである.筆者らによる受精卵でのノックアウトマウスの作製では,オフターゲットの候補として382箇所を調べて3箇所の切断であった11).マウスであれば,交配によりオフターゲットに変異をもたないマウスを選別することも可能であるし,統計処理による対応もできる.あるいは,同じ遺伝子を標的とするが異なるgRNAを用いて追試すれば,オフターゲットの切断による影響は無視できるであろう.
2本鎖DNA切断とDNA修復のエラーを利用したゲノム編集技術のメリットは遺伝子破壊にかぎらない.切断部位をオーバーラップする1本鎖オリゴDNAのなかほどに変異を導入しておけば,標的変異を内在性のゲノムに導入することができる.もちろん,単なるDNA切断に比べて変異の導入効率は低いが,ES細胞を使わずに受精卵から1世代で標的変異をもつマウスを作製できるのは魅力である.基本的には,両側に50塩基ほどの相同性をもつ100~130塩基のオリゴDNAを使うことにより,1塩基の置換やFLAGタグの挿入ができる(図3a).遺伝子を破壊すると致死となるような場合でも点変異の導入なら生存することも十分に考えられ,ヒトの疾患とまったく同じ変異を導入することなどで疾患モデルマウスとしての可能性が広がっている.ここでは,発想を逆転して,点変異の遺伝子治療に使われた例を紹介する.フマリルアセト酢酸ヒドラーゼをコードする遺伝子に変異をもつためチロシンを分解できず肝障害を起こす高チロシン血症モデルマウスに,この変異部位を切断するCas9およびgRNAを発現するプラスミドとともに,野生型の配列をもつ1本鎖DNAを静脈注射したところ,一部の肝細胞において遺伝子変異が正常にもどり,さらに,異常な細胞と徐々に置き換わることにより投薬治療の必要のなくなったことが報告された12).
数十塩基をこえるような長い配列を標的遺伝子座に導入したい場合には,1本鎖DNAではなく,相同性をもたせた2本鎖DNAを同時に注入することになる(図3a).ただし,2本鎖DNA切断により相同組換え率が改善されるとはいえ,受精卵への2本鎖DNAの注入により相同組換えマウスを作製することはむずかしい.およそ20%の初期胚においてレポーター遺伝子の発現が確認されたとの報告があるが13),筆者らの経験や私信では,実際に生まれてくる相同組換えマウスは多くても産仔の1~5%と考えられる.相同組換えマウスを確実に得るには1000個近い受精卵を処理して100匹近くを産ませる必要があり,コストや動物愛護の観点から,さらなる効率の改善がもとめられるであろう.現時点では,ES細胞における遺伝子ターゲティングのときに2本鎖DNA切断を誘導して効率化を図るほうが実用的かもしれない.
ところで,野生型のCas9ヌクレアーゼは2箇所の酵素活性部位をもち,2本鎖DNAの両方の鎖を別々に切断する.そこで,片方の活性部位を破壊したCas9 nickase(図5a),および,両方の活性部位を破壊したdead Cas9が作製されている(図5b).たとえば,Cas9 nickaseを2つのgRNAと組み合わせ,それぞれがセンス側およびアンチセンス側の両方を切断したときにはじめて遺伝子破壊を起こすことにより,オフターゲットの切断を軽減するというアプローチも報告されている14).また,dead Cas9を標的となる遺伝子座に結合させることにより転写制御の阻害をねらうCRISPRi法や15)(図5c),dead Cas9に修飾酵素をつないでエピゲノム修飾を改変する試み16)(図5d),あるいは,dead Cas9にGFPをつないで標的遺伝子を可視化する試みも報告されている17)(図5e).いずれもZFNやTALEを使って報告されてきたアプローチではあるが,gRNAを変えるだけでよいCRISPR/Cas系と組み合わせることで汎用性は飛躍的に改善される.まだ細胞レベルでの実験が多いため,今後は,個体レベルでの応用が期待される.また,2014年になり,Cas9ヌクレアーゼの立体構造も報告され18),原理面からのアプローチも可能となったことから,活性や特異性の改善にくわえて新たな利用法の開発もはじまっている.
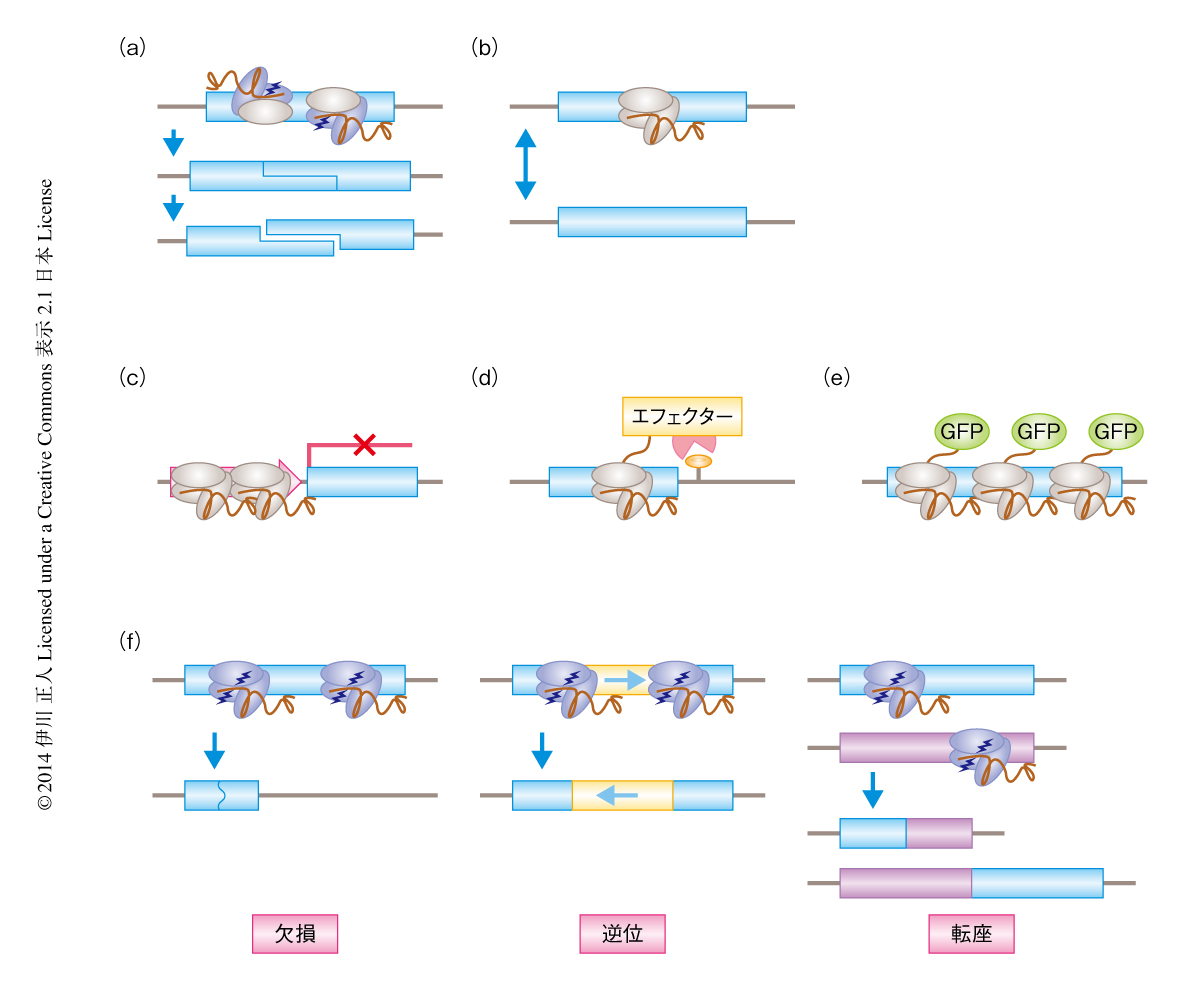
ここまでは,単一の遺伝子に対する遺伝子改変技術について紹介したが,もっとスケールの大きな遺伝子改変技術もある.これまで長い領域を欠損させる場合には,欠損させたい領域の両側にまたがる遺伝子ターゲティングベクターを用いて相同組換えを起こすことが多かった.あるいは,染色体の2箇所にloxP部位を挿入し,Creリコンビナーゼによりこれを抜き取るのが一般的であった.それがCRISPR/Cas法であれば,ゲノムの2箇所に対しgRNAをデザインしCas9ヌクレアーゼにより切断することでこれを簡単に実現できる(図5f).筆者らは60 kbpの領域を欠損させているが,私信ではMbpのレベルで欠損させたケースもある.いずれの場合も,解析したクローンの5~10%で欠損が生じるという実用可能な効率であった.また,同一の染色体の2箇所を切断すると一定の頻度で逆位を得ることもできるし,異なる染色体であれば転座を誘導することもできる(図5f).これらは,もはや染色体工学の域に入るであろう.
染色体工学といえば,人工染色体を生殖系列に伝達することのできるトランスクロモソミックマウスが開発されている19).染色体においてクラスターを構成するような薬物代謝関連酵素や,ヒト化抗体医薬の生産などに使える技術として注目されているが,いちどマウス化したのちの遺伝子改変が困難であった.ゲノム編集技術との組合せによる進展が期待される.
ところで,2倍体細胞では父方および母方の両方の対立遺伝子において遺伝子改変する必要があるが,1倍体細胞であれば1遺伝子の破壊あるいは改変により表現型が観察できる.2011年,マウスにおいて単為発生させた胚から1倍体のES細胞が樹立され,これを2倍体化すれば体細胞や生殖細胞に分化させることのできることが示された20).ヒトではKBM7細胞株やHAP1細胞株のように1倍体に近い状態で維持できる細胞株が知られており,ゲノム編集技術による大規模な遺伝子破壊が試みられている.1倍体のES細胞が安定的に維持できるようになれば活用が広がるであろう.
哺乳類細胞においてCRISPR/Cas法が機能することが報告されてから1年ほどのあいだに,ゲノム編集技術は遺伝子改変マウスの常識をくつがえし,生命科学および医学の研究に大きなインパクトをあたえた.予算面から本格的な活動にはいたっていないものの,わが国でも,CRISPR/Cas法を活用した次世代の遺伝子改変動物により先進的な医学研究を推進するプロジェクトが胎動している(URL:http://fmena.biken.osaka-u.ac.jp/).すでに,ラットやウサギ,家畜動物,サルでもゲノム編集技術の成功が報告されており,疾患モデルの開発にくわえ治療法への応用など,今後は前臨床研究の推進による臨床研究あるいは創薬への橋渡しも期待される.
略歴:1997年 大阪大学大学院薬学研究科博士課程 修了,2000年 米国The Salk Institute博士研究員,2004年 大阪大学微生物病研究所 助教授を経て,2011年より同 教授.
抱負:新しい遺伝子改変技術を開発および応用しながら,受精を中心とする哺乳類の生殖の機構を明らかにしたい.
研究室URL:http://www.egr.biken.osaka-u.ac.jp/
© 2014 伊川 正人 Licensed under CC 表示 2.1 日本
(大阪大学微生物病研究所 附属感染動物実験施設)
email:伊川正人
領域融合レビュー, 3, e008 (2014) DOI: 10.7875/leading.author.3.e008
Masahito Ikawa: Genome editing in mice opens a new era for biological and biomedical researches.
要 約
遺伝子改変動物を用いた研究の醍醐味は,培養細胞や試験管内ではみられないダイナミックな高次生命現象を,個体レベルで観察あるいは解析できることにある.とくに,ヒトと同じ哺乳類に属するマウスは,全ゲノム塩基配列が明らかにされているだけでなく,ES細胞における相同組換えを利用した標的遺伝子の破壊が可能であることから,遺伝子機能解析ツールとしてだけでなく,ヒトの疾患の病態モデルとして,生命科学研究および医学研究の礎を担っている.さらに最近では,黒船襲来ともいうべきゲノム編集技術,とくにRNAにより標的配列を認識するCRISPR/Cas法の開発により,1塩基レベルから染色体レベルでの遺伝子の挿入,欠損,置換,さらには,エフェクターとの組合せによる転写制御,ゲノムの可視化,エピゲノム修飾の操作などの可能性が大きく広がっている.このレビューでは,従来の遺伝子改変マウスの作製技術をふまえ,ゲノム編集がひらく遺伝子改変マウスの未来について解説する.
はじめに
マウスは約20~30 gと小さく穏やかな性質をもち多産な動物である.また,性成熟(雄で約6週間,雌で約4週間)や妊娠期間(約20日間)を含めて比較的ライフサイクルが短いことから,古くから実験動物として供されてきた.さらに,全ゲノム塩基配列の解読によりヒトの遺伝子の99%はマウスにも保存されていることが明らかにされたこと,体外受精や胚操作などの生殖工学的な手法も確立されていることなどもあり,マウスは遺伝学的にも微生物学的にも制御されたすばらしい実験動物であるといえる.
個体レベルにおいてマウスのゲノム遺伝子を操作する試みは古く,さまざまな手法が活用されてきた.もっとも古典的な方法は自然変異マウスのスクリーニングであり,胸腺を欠損するヌードマウスや,T細胞およびB細胞を欠損するSCIDマウスなどが有名である.つづいて,放射線の照射や化学変異原の投与などの人為突然変異法が開発された.たとえば,エチルニトロソ尿素(ethylnitrosourea:ENU)の投与は自然変異の約100倍~1000倍の確率で点変異を誘発し,ヌル欠損変異だけでなくアミノ酸置換変異なども得られる.方法は簡単で,エチルニトロソ尿素を投与した雄と野生型の雌を交配させて得られる子孫を表現型によりスクリーニングする.しかしながら,ゲノムにおいて変異を同定することは非常に困難であった.近年の塩基配列解析技術の飛躍的な進展にともない,ランダムに導入された変異をさきにスクリーニングするというアプローチも可能にはなっているが1),思いどおりの変異を導入できないという難点は残る.なお,自然変異マウスおよび人為突然変異マウスはいずれも遺伝子組換え実験の規制にとらわれず,野生型のマウスと同様に扱うことができる.
変異マウスを用いて表現型からその原因となる遺伝子を探りあてる順遺伝学(フォワードジェネティクス)に対し,遺伝子を改変して表現型との関連を探るアプローチを逆遺伝学(リバースジェネティクス)という.1970年代に外来の遺伝子を導入したトランスジェニックマウスが,1980年台後半に内在性の遺伝子を破壊したノックアウトマウスが開発されたことにより,逆遺伝学は大きく進展した.さらに2010年台に入り,ゲノム編集技術を活用した遺伝子改変マウスの可能性が大きく広がっている.以下,遺伝子組換えマウスの作製技術とその可能性について解説する.
1.トランスジェニックマウス
一般的には,トランス遺伝子とよばれる外来性の遺伝子を宿主のゲノムに人為的に組み込み,これが世代をこえて受け継がれるマウスをさす.宿主がもたないタンパク質を発現させたり,野生型あるいは変異型のタンパク質を過剰に発現させたりする目的で作製されることが多い.たとえば,筆者らが開発した蛍光タンパク質EGFPを発現するグリーンマウスは移植やがん転移など幅広い基礎研究に,また,rasH2マウスは発がん試験に用いられている.組織特異的なプロモーターや薬剤誘導型のプロモーターを使うことにより,より複雑な遺伝子発現の制御も可能となる.
トランスジェニックマウスは受精卵の前核にガラス管を用いて直鎖状のDNAを直接注入することにより作製することが多い(図1a).トランス遺伝子はゲノムの1箇所にタンデムに複数のコピーが組み込まれることが多く,挿入位置やコピー数により発現パターンや強度が左右される.数は少ないが,レンチウイルスベクターを受精卵に感染させる手法も使われる(図1b).この場合は,ゲノムの複数の個所に1コピーずつ組み込まれるため,位置効果を相殺することができる.また,前核に注入する方法に比べ作製効率が10倍近く改善されることから,稀少な変異マウス系統を使うときや第1世代での発現解析などに威力を発揮する.
なお,従来の方法では胎仔と胎盤の両方に遺伝子が導入されるため,胚性致死などの解析が困難であった.筆者らは,発生の進んだ胚盤胞期の胚にレンチウイルスベクターを感染させれば,胎仔に遺伝子導入することなく胎盤に特異的に遺伝子導入することができることを見い出した(図1c).妊娠期の遺伝子機能解析ツールとして活用が期待される.
2.ノックアウトマウスの開発
ノックアウトマウスは内在性の遺伝子が破壊されたマウスの総称として使われており,点変異によるフレームシフト,ナンセンス変異,ミスセンス変異による機能の欠失,のちに述べるゲノム編集技術などにより遺伝子を破壊されたマウスを含む.一般的には,内在性の遺伝子を破壊したES細胞(embryonic stem cell,胚性幹細胞)からキメラマウスを介して生殖系列に変異した遺伝子を伝達するマウスをさすことが多い.
1981年,マウスの胚盤胞からES細胞が樹立され,胚盤胞に混合すればキメラマウスが作製できること,さらに,交配によりES細胞に由来する子孫を得られることが報告された.そののち,薬剤により選択することのできる培養細胞株の利点を生かし,ES細胞を用いたマウスの遺伝子破壊法として有名な2つの方法が開発された.ランダムに遺伝子を破壊する遺伝子トラップ法と,標的とした遺伝子を破壊する遺伝子ターゲティング法である.遺伝子トラップ法は,外来性の薬剤耐性遺伝子がゲノムにランダムに挿入される際に,偶然に内在性の遺伝子座に挿入されたものを薬剤により選択する.遺伝子ターゲティング法は,外来性の薬剤耐性遺伝子の両側にゲノムと相同性をもつ領域を付加し,相同組換えにより内在性の遺伝子を薬剤耐性遺伝子と置き換える方法であり,目的とする遺伝子の機能を個体レベルで解析する逆遺伝学の典型例とされる.1980年台の終わりから1990年代にかけて,ノックアウトマウスに関する多くの論文がトップジャーナルに掲載され,個体レベルでの遺伝子の機能解析にはノックアウトマウスが常道とされるようになった.2007年には,ノックアウトマウスの開発者であるMartin Evans,Oliver Smithies,Mario Capecchiにノーベル医学生理学賞が授与されている.
3.遺伝子トラップ法
遺伝子トラップ法においては,当初は薬剤耐性遺伝子とポリA付加シグナルをつないだカセットを用いて内在性のプロモーターをとらえるプロモータートラップ法が主流であったが,ES細胞で発現している遺伝子しか破壊できないという欠点があった(図2a).そこで考案されたのが,普遍的に発現するプロモーターの後ろにレポーター遺伝子を挿入したカセットを用いてポリAをとらえるポリAトラップ法で,原理的にはすべての遺伝子をトラップして破壊できる.しかし,ポリAトラップ法では薬剤耐性遺伝子のもつ終止コドンの後ろに内在性のエキソンが連なるため,ナンセンス変異依存性mRNA分解機構(non-sense mediated mRNA decay:NMD)によりmRNAの不安定化をひき起こすという問題があった.現在では,薬剤耐性遺伝子の後ろにリボソームエントリー部位と3フレームのATGを付加することなどで問題は解決されている2).遺伝子トラップカセットの導入には,直線化プラスミドのエレクトロポレーション法も利用されるが,細胞の生存率や導入効率からレトロウイルスベクターを使った方法が好まれる.最近は,Sleeping BeautyやPiggyBacなどのトランスポゾン系を使ってゲノムに組み込むアプローチも開発されており,その場合は,表現型を観察したのちにトラップベクターを抜き取って機能の回復を確認できるという利点がある(図2a).
4.遺伝子ターゲティング法
遺伝子トラップ法は同じベクターをくり返しES細胞に導入することによりランダムに遺伝子を破壊するのに対し,遺伝子ターゲティング法では標的とする遺伝子ごとにデザインされたベクターを作製してES細胞へ導入し,薬剤耐性遺伝子および薬剤感受性遺伝子(あるいは,自殺遺伝子)を組み合わせた正の選択および負の選択により相同組換えを起こしたES細胞を濃縮する(図2b).つぎに,遺伝子を導入したES細胞を薬剤により選択しクローン化する(図2c).クローン化したES細胞からゲノムDNAを抽出してPCR法やサザンブロット法により相同組換えを確認する.ES細胞を初期胚と凝集し,偽妊娠マウスに移植してキメラマウスを作製する(図2d).当初,汎用された129系統のES細胞はC57BL/6系統の胚盤胞期の胚に注入してキメラマウスを作製していたが,C57BL/6系統のES細胞は,ICR系統の8細胞期の胚と凝集させるほうが生殖系列に寄与しやすいようである.得られたキメラマウスを野生型マウスと交配させて,標的遺伝子に変異をもつF1マウスを得たのち,ヘテロF1マウスの雌雄を交配してホモ変異をもつF2マウスを得る(図2e).ベクターの構築から,ES細胞における相同組換え,キメラマウスの作製と交配による生殖系列への寄与,さらに,交配によるホモ欠損マウスを得るためには,年単位の実験になることも多い.
マウスではES細胞の培養技術,遺伝子改変技術,キメラ動物の作製技術が確立しているが,ほかの動物種ではそうともかぎらない.たとえばラットでは,マウスから遅れること15年,2003年になってようやくES細胞を使った遺伝子ノックアウトが報告された.ES細胞以外の細胞を用いた遺伝子ターゲティング法も開発されている.iPS細胞を用いればES細胞と同様の実験ができることはいうまでもないが,GS細胞(germ line stem cell,雄性生殖幹細胞)において標的遺伝子を破壊し,それを精巣に移植して精子を形成させ次世代を得ることもできる3).なお,家畜動物などでは,遺伝子ターゲティングした胎仔繊維芽細胞からクローン技術により個体を復元する方法も一般的である.
遺伝子ノックアウト実験の醍醐味は,表現型をみるまで標的遺伝子の重要性がわからないことであろう.筆者らもこれまで数多くのノックアウトマウスを作製してきたが,in vitroにおける実験の結果から誰もが必須と信じて疑わなかった遺伝子が不要であったり,まったく予期せぬ結果から生命現象に必須の遺伝子がみつかったりしたケースも少なくない.ノックアウトマウスの結果しだいでは定説を一発逆転できることから,予想どおりの表現型より,むしろ予想外の結果が得られたほうが大きな発見につながる.
5.国際ノックアウトマウスプロジェクト
遺伝子ターゲティングの際の重要なポイントは,生殖系列に寄与できる未分化な状態を維持したES細胞の培養と,効率的な相同組換えである.ES細胞の培養については,由来するマウスの系統や樹立されたES細胞株などによって異なり,培地や血清のロットにも左右される.そのため,遺伝子操作と継代の過程において性質が変化してキメラマウスが得られなかったり,キメラマウスが誕生しても遺伝子改変したES細胞に由来する仔が得られず,実験が振り出しにもどるケースも少なくなかった.効率的な相同組換えについては,組換えの効率を上げる目的で相同領域を長くとるため(両側合計で7~10 kbp),全長が20 kbp近いプラスミドの切り貼りとなり,ベクターの構築と配列の確認に手間と時間を要するという問題点があった.また,相同組換えの頻度は標的遺伝子座やベクターのもつ相同領域の配列などによっても大きく異なるため,トラブルシューティングは経験や勘にたよる部分も多く,運が悪いのか腕が悪いのかわからないなど初心者にはむずかしい実験であった.
さらに残念なことに,コストや労力をかけて作製したがゆえに,得られたノックアウトマウスを研究材料として共有しない研究室もあり,有名な遺伝子のノックアウトマウスは多くの研究室で重複して作製された.また,表現型のみられないノックアウトマウスの情報は表にでないため,同じノックアウトマウスが異なる研究室でくり返し作製されるといったむだも生じていた.
これらの背景から,少ない拠点においてすべての遺伝子を網羅的にノックアウトして人類共通の財産にしようという国際ノックアウトマウスプロジェクトが始動した4).米国やヨーロッパを中心に活動が広がり,2006年ごろ,IKMC(International Knockout Mouse Consortium)が国際プロジェクトとして活動を本格化した.当初は遺伝子トラップ法が主流であったが,相同組換え率の高いベクターの開発や,ベクター構築の効率化あるいは機械化により,遺伝子ターゲティング法も採用されている.IKMCで採用された遺伝子ターゲティングベクターの多くが組織特異的あるいは時期特異的に遺伝子を破壊できるコンディショナルノックアウトタイプであり,内在性のプロモーターによりレポーターとしてLacZ遺伝子を発現するといったくふうもなされている.
なお,2011年から国際ノックアウトマウスプロジェクトはIMPC(International Mouse Phenotyping Consortium,URL:http://www.mousephenotype.org/)にひき継がれ,これまでに,IKMCとあわせて2万近い遺伝子について組換えES細胞株が樹立されている.今後は,キメラマウスを介したノックアウトマウスの作製と1次的な表現型の解析が主体となり,解析の結果は世界中に公開される.当初5年で4000系統の解析を終えるという目標が設定されているが,細胞レベルとは異なり個体レベルの実験には莫大な費用と時間がかかる.2014年6月時点では約300系統の表現型の公開にとどまっており,今後の進展が期待される.
6.ゲノム編集技術と人工制限酵素
欧米各国が莫大な予算を投じて国際ノックアウトマウスプロジェクトを推進するかたわら,新たな遺伝子改変技術が産声をあげていた.人工制限酵素の開発と応用による標的遺伝子の改変技術,つまり,ゲノム編集技術である.原理的には,ゲノムの2本鎖DNAを切断し,その修復機構のエラーを利用して遺伝子を効率的に改変する(図3a).非相同末端結合(nonhomologous end-joining:NHEJ)経路による修復では,切断をうけたDNA末端を直接つなぎ合わせる際に,数塩基の欠失あるいは挿入などのエラーが起こりやすい.また,相同組換え(homologous recombination:HR)経路による修復ではエラーは少ないが,相同性をもつ1本鎖DNAあるいは2本鎖DNAを人為的に導入しておくとあやまってゲノムに取り込まれることがある.ゲノム編集技術では,このような2本鎖DNA切断とその修復のエラーをうまく利用して遺伝子改変を行う5).
ゲノム編集といえばZFN(zinc finger nuclease)が最初と思う人が多いかもしれないが,それ以前から,2本鎖DNA切断を利用した遺伝子改変は試みられていた.1996年には,18塩基を特異的に認識するI-SceIメガヌクレアーゼを用いた2本鎖DNA切断により相同組換えの効率の改善することが報告されている.しかし,メガヌクレアーゼは標的配列をデザインすることができないため,認識配列をさきにゲノムに組み込む必要があるなど,実用的にはほとんど意味のないものであった.同じく1996年に開発されたのがZFNである.ジンクフィンガーモチーフとよばれるDNA結合ドメインとFokIヌクレアーゼで構成された人工キメラタンパク質であり,標的遺伝子を認識し切断する(図3b).しかし,ZFNではDNA結合ドメインがデザインどおりにDNAを認識しないことが多く,ベクターの構築がむずかしいとの欠点があった.また,市販品が非常に高額であったため,一般の研究者には高嶺の花であることも多かった.
つぎに登場したのが,ZFNのDNA結合ドメインを植物病原細菌であるXanthomonas属細菌のもつTALEエフェクターに代えたTALENである(図3c).核酸のA,C,G,Tそれぞれ1塩基に特異的に対応するユニットを標的配列にあわせて組み合わせるだけで構築できることから,一般の研究者にも利用できるレベルになった.さらに数塩基を認識するモジュールを組み合わせる改善もなされている.また,34アミノ酸残基からなるユニットの4番目と32番目のアミノ酸残基のバリエーションに注意してモジュールを構築することにより,活性の高いTALENを効率よく作製できるPlatinum TALENも開発されている6)(URL:http://www.addgene.org/TALEN/PlatinumGate/).
ZFNやTALENといった人工制限酵素は,受精卵に注入するだけで内在性の遺伝子を破壊できることから,ホヤ,カエル,ゼブラフィッシュなど,多くのモデル生物において利用されている.詳細については末尾にあげた参考図書を参照されたい.
7.CRISPR/Cas法によるゲノム編集技術
ZFNやTALENの登場により“人工制限酵素”や“ゲノム編集”という言葉が研究者の耳目をあつめ,また,画期的な遺伝子改変技術としての評価から,2011年にはNature Method誌のMethod of the Year,2012年にはScience誌のBreakthrough of the yearにとりあげられた.そのような状況のなか,次世代のゲノム編集技術として開発されたのがCRISPR/Cas法である.そもそも,CRISPR/Cas系は古細菌などがもつ獲得免疫機構であり,ファージなどを介して侵入する外来性のDNAを断片化して自らのゲノムに組み込み,2度目の感染のときに,この外来性のDNAの断片から発現する2つの小分子RNAを用いて標的DNAを認識し,Cas9ヌクレアーゼをリクルートしてこれを切断する.2013年2月には,2つの小分子RNAをひとつのキメラRNAとし(guide RNA:gRNA,あるいは,single guide RNA:sgRNAとよばれる),Cas9との2つの因子だけでゲノムDNAの切断活性を発揮できることが示された7,8)(図3d).
ZFNやTALENはDNA結合ドメインと切断ドメインを融合タンパク質として機能させるため標的遺伝子ごとに複雑なベクターの構築が必要なのに対し,CRISPR/Cas法では標的配列に対応するgRNAの5’末端の20塩基を変更するだけで標的遺伝子を変えることができる.この手軽さと低コスト,高い切断効率と再現性から,CRISPR/Cas法は瞬く間に生命科学研究者に広まった.筆者らはプラスミドpX330(URL:http://www.addgene.org/42230/)を用いているが,pX330にはgRNA発現カセットおよびコドンをヒト化したCas9発現カセットが搭載されており,合成オリゴDNAを導入するだけでノックアウトベクターを構築できる(図4a).
CRISPR/Cas法が哺乳類培養細胞において機能することが示されてから半年も経たない2013年5月,Cas9をコードするmRNAとgRNAを受精卵の細胞質に注入することによりノックアウトマウスが得られるという論文が報告された9)(図4b).当時,筆者らも,RNA合成のステップをスキップしてpX330プラスミドを受精卵の前核にマイクロインジェクションする簡易法を独自に開発していたが,2番手の論文ということで予想以上に審査が厳しく,論文発表までに半年を要した10).CRISPR/Cas法を用いたノックアウトマウス作製のメリットはその簡便性とスピードであり,実際に,合成オリゴDNAの発注からpX330ベクターの構築,培養細胞における切断活性の検討に約1週間,それに受精卵への注入から出産までの3週間をくわえた約1カ月でノックアウトマウスが作製できる.さらに,変異マウスの約半数において両方の対立遺伝子に変異が導入されている点は特筆すべきであり,ES細胞を使うと交配だけでも半年を要することを考えると,ファウンダー世代でホモ欠損マウスが得られるのは夢のようである.筆者らの経験では,100個の受精卵を処理すれば約10匹が生まれ,5匹程度が変異マウス,うち2匹程度が両方の対立遺伝子に変異をもつ10,11).なお,受精卵ではなく,ES細胞へのプラスミドのトランスフェクションでは,pX330が一過性に導入されたES細胞の約8割で両方の対立遺伝子に変異が認められる.
CRISPR/Cas法の注意点としては,標的とする配列に近似した配列(オフターゲット)を切断してしまうリスクがある.ZFNやTALENでは,センス鎖とアンチセンス鎖を認識する2つのユニットが同時に認識してはじめてFokIによる切断が起こるのに対し,片側のDNA鎖のみを認識するCRISPR/Cas法の特異性は低いとされ,がん細胞などの培養細胞などではオフターゲットの切断が多く報告されている.しかし,受精卵やES細胞などではDNA修復能力が高いのか,それほど問題にならないようである.筆者らによる受精卵でのノックアウトマウスの作製では,オフターゲットの候補として382箇所を調べて3箇所の切断であった11).マウスであれば,交配によりオフターゲットに変異をもたないマウスを選別することも可能であるし,統計処理による対応もできる.あるいは,同じ遺伝子を標的とするが異なるgRNAを用いて追試すれば,オフターゲットの切断による影響は無視できるであろう.
8.ゲノム編集技術の未来
2本鎖DNA切断とDNA修復のエラーを利用したゲノム編集技術のメリットは遺伝子破壊にかぎらない.切断部位をオーバーラップする1本鎖オリゴDNAのなかほどに変異を導入しておけば,標的変異を内在性のゲノムに導入することができる.もちろん,単なるDNA切断に比べて変異の導入効率は低いが,ES細胞を使わずに受精卵から1世代で標的変異をもつマウスを作製できるのは魅力である.基本的には,両側に50塩基ほどの相同性をもつ100~130塩基のオリゴDNAを使うことにより,1塩基の置換やFLAGタグの挿入ができる(図3a).遺伝子を破壊すると致死となるような場合でも点変異の導入なら生存することも十分に考えられ,ヒトの疾患とまったく同じ変異を導入することなどで疾患モデルマウスとしての可能性が広がっている.ここでは,発想を逆転して,点変異の遺伝子治療に使われた例を紹介する.フマリルアセト酢酸ヒドラーゼをコードする遺伝子に変異をもつためチロシンを分解できず肝障害を起こす高チロシン血症モデルマウスに,この変異部位を切断するCas9およびgRNAを発現するプラスミドとともに,野生型の配列をもつ1本鎖DNAを静脈注射したところ,一部の肝細胞において遺伝子変異が正常にもどり,さらに,異常な細胞と徐々に置き換わることにより投薬治療の必要のなくなったことが報告された12).
数十塩基をこえるような長い配列を標的遺伝子座に導入したい場合には,1本鎖DNAではなく,相同性をもたせた2本鎖DNAを同時に注入することになる(図3a).ただし,2本鎖DNA切断により相同組換え率が改善されるとはいえ,受精卵への2本鎖DNAの注入により相同組換えマウスを作製することはむずかしい.およそ20%の初期胚においてレポーター遺伝子の発現が確認されたとの報告があるが13),筆者らの経験や私信では,実際に生まれてくる相同組換えマウスは多くても産仔の1~5%と考えられる.相同組換えマウスを確実に得るには1000個近い受精卵を処理して100匹近くを産ませる必要があり,コストや動物愛護の観点から,さらなる効率の改善がもとめられるであろう.現時点では,ES細胞における遺伝子ターゲティングのときに2本鎖DNA切断を誘導して効率化を図るほうが実用的かもしれない.
ところで,野生型のCas9ヌクレアーゼは2箇所の酵素活性部位をもち,2本鎖DNAの両方の鎖を別々に切断する.そこで,片方の活性部位を破壊したCas9 nickase(図5a),および,両方の活性部位を破壊したdead Cas9が作製されている(図5b).たとえば,Cas9 nickaseを2つのgRNAと組み合わせ,それぞれがセンス側およびアンチセンス側の両方を切断したときにはじめて遺伝子破壊を起こすことにより,オフターゲットの切断を軽減するというアプローチも報告されている14).また,dead Cas9を標的となる遺伝子座に結合させることにより転写制御の阻害をねらうCRISPRi法や15)(図5c),dead Cas9に修飾酵素をつないでエピゲノム修飾を改変する試み16)(図5d),あるいは,dead Cas9にGFPをつないで標的遺伝子を可視化する試みも報告されている17)(図5e).いずれもZFNやTALEを使って報告されてきたアプローチではあるが,gRNAを変えるだけでよいCRISPR/Cas系と組み合わせることで汎用性は飛躍的に改善される.まだ細胞レベルでの実験が多いため,今後は,個体レベルでの応用が期待される.また,2014年になり,Cas9ヌクレアーゼの立体構造も報告され18),原理面からのアプローチも可能となったことから,活性や特異性の改善にくわえて新たな利用法の開発もはじまっている.
9.そのほかの技術
ここまでは,単一の遺伝子に対する遺伝子改変技術について紹介したが,もっとスケールの大きな遺伝子改変技術もある.これまで長い領域を欠損させる場合には,欠損させたい領域の両側にまたがる遺伝子ターゲティングベクターを用いて相同組換えを起こすことが多かった.あるいは,染色体の2箇所にloxP部位を挿入し,Creリコンビナーゼによりこれを抜き取るのが一般的であった.それがCRISPR/Cas法であれば,ゲノムの2箇所に対しgRNAをデザインしCas9ヌクレアーゼにより切断することでこれを簡単に実現できる(図5f).筆者らは60 kbpの領域を欠損させているが,私信ではMbpのレベルで欠損させたケースもある.いずれの場合も,解析したクローンの5~10%で欠損が生じるという実用可能な効率であった.また,同一の染色体の2箇所を切断すると一定の頻度で逆位を得ることもできるし,異なる染色体であれば転座を誘導することもできる(図5f).これらは,もはや染色体工学の域に入るであろう.
染色体工学といえば,人工染色体を生殖系列に伝達することのできるトランスクロモソミックマウスが開発されている19).染色体においてクラスターを構成するような薬物代謝関連酵素や,ヒト化抗体医薬の生産などに使える技術として注目されているが,いちどマウス化したのちの遺伝子改変が困難であった.ゲノム編集技術との組合せによる進展が期待される.
ところで,2倍体細胞では父方および母方の両方の対立遺伝子において遺伝子改変する必要があるが,1倍体細胞であれば1遺伝子の破壊あるいは改変により表現型が観察できる.2011年,マウスにおいて単為発生させた胚から1倍体のES細胞が樹立され,これを2倍体化すれば体細胞や生殖細胞に分化させることのできることが示された20).ヒトではKBM7細胞株やHAP1細胞株のように1倍体に近い状態で維持できる細胞株が知られており,ゲノム編集技術による大規模な遺伝子破壊が試みられている.1倍体のES細胞が安定的に維持できるようになれば活用が広がるであろう.
おわりに
哺乳類細胞においてCRISPR/Cas法が機能することが報告されてから1年ほどのあいだに,ゲノム編集技術は遺伝子改変マウスの常識をくつがえし,生命科学および医学の研究に大きなインパクトをあたえた.予算面から本格的な活動にはいたっていないものの,わが国でも,CRISPR/Cas法を活用した次世代の遺伝子改変動物により先進的な医学研究を推進するプロジェクトが胎動している(URL:http://fmena.biken.osaka-u.ac.jp/).すでに,ラットやウサギ,家畜動物,サルでもゲノム編集技術の成功が報告されており,疾患モデルの開発にくわえ治療法への応用など,今後は前臨床研究の推進による臨床研究あるいは創薬への橋渡しも期待される.
文 献
- Gondo, Y., Fukumura, R., Murata, T. et al.: ENU-based gene-driven mutagenesis in the mouse: a next-generation gene-targeting system. Exp. Anim., 59, 537-548 (2010)[PubMed]
- Shigeoka, T., Kawaichi, M. & Ishida, Y.: Suppression of nonsense-mediated mRNA decay permits unbiased gene trapping in mouse embryonic stem cells. Nucleic Acids Res., 33, e20 (2005)[PubMed]
- Kanatsu-Shinohara, M., Ikawa, M., Takehashi, M. et al.: Production of knockout mice by random or targeted mutagenesis in spermatogonial stem cells. Proc. Natl. Acad. Sci. USA, 103, 8018-8023 (2006)[PubMed]
- Austin, C. P., Battey, J. F., Bradley, A. et al.: The knockout mouse project. Nat. Genet., 36, 921-924 (2004)[PubMed]
- Gaj, T., Gersbach, C. A. & Barbas, C. F. 3rd.: ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol., 31, 397-405 (2013)[PubMed]
- Sakuma, T., Ochiai, H., Kaneko, T. et al.: Repeating pattern of non-RVD variations in DNA-binding modules enhances TALEN activity. Sci. Rep., 3, 3379 (2013)[PubMed]
- Cong, L., Ran, F. A., Cox, D. et al.: Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819-823 (2013)[PubMed]
- Mali, P., Yang, L., Esvelt, K. M. et al.: RNA-guided human genome engineering via Cas9. Science, 339, 823-826 (2013)[PubMed]
- Wang, H., Yang, H., Shivalila, C. S. et al.: One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell, 153, 910-918 (2013)[PubMed]
- Mashiko, D., Fujihara, Y., Satouh, Y. et al.: Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci. Rep., 3, 3355 (2013)[PubMed]
- Mashiko, D., Young, S. A., Muto, M. et al.: Feasibility for a large scale mouse mutagenesis by injecting CRISPR/Cas plasmid into zygotes. Dev. Growth Differ., 56, 122-129 (2014)[PubMed]
- Yin, H., Xue, W., Chen, S. et al.: Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol., 32, 551-553 (2014)[PubMed]
- Yang, H., Wang, H., Shivalila, C. S. et al.: One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell, 154, 1370-1379 (2013)[PubMed]
- Ran, F. A., Hsu, P. D., Lin, C. Y. et al.: Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell, 154, 1380-1389 (2013)[PubMed]
- Qi, L. S., Larson, M. H., Gilbert, L. A. et al.: Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell, 152, 1173-1183 (2013)[PubMed]
- Perez-Pinera, P., Kocak, D. D., Vockley, C. M. et al.: RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods, 10, 973-976 (2013)[PubMed]
- Chen, B., Gilbert, L. A., Cimini, B. A. et al.: Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell, 155, 1479-1491 (2013)[PubMed]
- Nishimasu, H., Ran, F. A., Hsu, P. D. et al.: Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell, 156, 935-949 (2014)[PubMed][新着論文レビュー]
- Kazuki, Y., Hoshiya, H., Takiguchi, M. et al.: Refined human artificial chromosome vectors for gene therapy and animal transgenesis. Gene Ther., 18, 384-393 (2011)[PubMed]
- Leeb, M. & Wutz, A.: Derivation of haploid embryonic stem cells from mouse embryos. Nature, 479, 131-134 (2011)[PubMed]
参考図書
山本 卓 (編): 今すぐ始めるゲノム編集 (実験医学別冊). 羊土社 (2014)
Yamamoto, T. & Nakamura, H. (eds.): Special Issue: Genome Editing. Dev. Growth Differ., 56, 1-129 (2014)
著者プロフィール
略歴:1997年 大阪大学大学院薬学研究科博士課程 修了,2000年 米国The Salk Institute博士研究員,2004年 大阪大学微生物病研究所 助教授を経て,2011年より同 教授.
抱負:新しい遺伝子改変技術を開発および応用しながら,受精を中心とする哺乳類の生殖の機構を明らかにしたい.
研究室URL:http://www.egr.biken.osaka-u.ac.jp/
© 2014 伊川 正人 Licensed under CC 表示 2.1 日本
