自己炎症性疾患の病態および分子機構
2014/11/21
安友 康二
(徳島大学大学院ヘルスバイオサイエンス研究部 生体防御医学分野)
email:安友康二
領域融合レビュー, 3, e013 (2014) DOI: 10.7875/leading.author.3.e013
Koji Yasutomo: Molecular basis of autoinflammatory disorders.

自己抗原に応答するリンパ球がその発症の鍵となる自己免疫疾患が古くから免疫難病として知られている.それと対比される疾患として,自己炎症性疾患とよばれる疾患が家族性の炎症性疾患における原因遺伝子の研究から確立されてきた.自己炎症とは,自己免疫応答や明らかな感染が存在しないにもかかわらず炎症が誘導される病態であり,非常に幅広い疾患の基盤になると考えられる.最近のゲノム解析手法の発展により,自己炎症性の病態を誘導する遺伝子について解明が進みつつあり,NLRP3に代表されるNOD様受容体ファミリーの遺伝子異常にくわえ,最近,免疫プロテアソームの構成タンパク質の遺伝子異常による自己炎症性疾患が報告された.これらの遺伝子が単一因子性の疾患にとどまらず多因子性の疾患にも関与することも報告されつつあり,今後,自己炎症に関与する疾患はますます明らかになると予測される.それと並行して,自己炎症性の病態に焦点をあてた治療法の開発が今後の研究の中心的な課題になると思われる.
炎症という病態の概念は非常に広範囲であり,炎症は古くから多くの疾患に寄与していると考えられてきた.一方で,炎症のかかわる疾患は多岐にわたるにもかかわらず,その分子機序の詳細はながらく明らかにされていなかった.ところが,家族性の炎症性疾患における原因遺伝子の発見をひとつの契機として1,2),炎症を誘導する分子機構の詳細がひもとかれつつある3).また同時に,悪性腫瘍や神経変性疾患などのように,これまで炎症との関連性が明確ではなかった疾患と炎症との関連性についても報告されるようになってきた4).以上のような背景から,炎症はこれまで考えられてきた以上に幅広い疾患の基盤となり,病態の進展に関与していると考えられるようになってきた.その考えにもとづき,現代の医療においては,どのように炎症を制御するかということが治療を考えるうえで中心的な課題になろうとしている.
炎症は内因性の異常,感染,毒物など外因性の物質などにより誘導されると考えられ,刺激に依存して発動される細胞内シグナル伝達経路やひき起こされる炎症の病態も多様である.免疫系の異常により誘導される疾患として古くから自己免疫疾患の概念が確立されており,自己免疫疾患は自己抗原を認識するT細胞およびB細胞が過剰に活性化あるいは増殖することにより発症すると考えられている.一方,最近の研究から,自己免疫応答や明らかな感染が存在しないにもかかわらず,自然免疫系を制御する細胞の過剰な活性化を契機とする激しい炎症を特徴とする疾患が見い出され,それらは自己炎症性疾患と定義されその概念が確立されつつある1,5).自己炎症性疾患は,狭義には単一の遺伝子の異常により発症する遺伝性の疾患をさすことが多いが,遺伝性が明らかではない疾患,あるいは,多因子性に発症すると考えられている疾患のなかにも自己炎症性疾患として分類の可能な疾患があると思われる.さらに,自己炎症性疾患を誘導するシグナル伝達経路はその進展の過程に寄与していると考えられることから,自己炎症は多岐にわたる疾患に寄与しているといえる(図1).
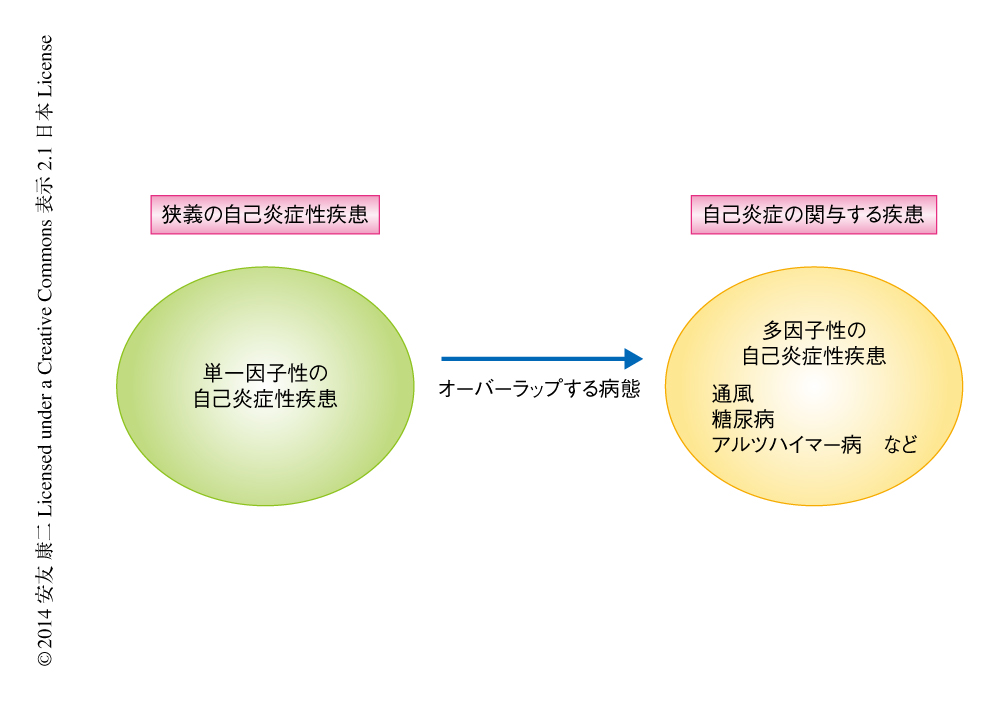
自己炎症性疾患の研究の進展は,NLRP3などの単一の因子の異常に起因する疾患の分子病態の発見が契機となり発展してきた1,2,6-11).そのような単一の遺伝子変異による疾患に関する研究から派生して,最近,NLRP3は糖尿病あるいはアルツハイマー病の病態にも寄与することが明らかになりつつある.このレビューでは,自己炎症性疾患の発症に寄与する分子の詳細とその病態について解説する.
インフラマソームは細胞質に侵入した病原性微生物あるいは尿酸などの結晶により活性化するタンパク質複合体の総称である.一般的には,インフラマソームの活性化によりカスパーゼ1前駆体が活性をもつカスパーゼ1に変換し,インターロイキン1βやインターロイキン18などの炎症性サイトカインの産生を誘導することにより炎症を惹起することが知られている5).インフラマソームを構成するタンパク質としてはNOD様受容体が知られている.NOD様受容体ファミリーは細胞に感染する微生物を認識するタンパク質として進化の過程において多様性を獲得してきたと推測される.ここでは,NOD様受容体ファミリーのうち,自己炎症性疾患との関連性が深いNLRP3について解説する.
NLRP3インフラマソームはβアミロイド,尿酸の結晶,細胞外のATPにより活性化されることが知られている12,13).しかし,それらの分子がどのような分子機序によりNLRP3インフラマソームを活性化するかについての詳細はいまだ理解されていない.しかし,K+の流出やリソソーム膜の損傷などの機序が考えられており,さらに最近では,ミトコンドリアの関与などいくつかの重要な知見が報告されている.K+の関与に関しては,K+が細胞外に流出することにより細胞内のK+濃度が低下しNLRP3インフラマソームが活性化することが示されている14).実際に,K+の流出を抑制するとNLRP3インフラマソームの活性化が抑制されることも明らかにされている.高濃度のATPはNLRP3インフラマソームを活性化させるが,これはATPによりP2X7受容体を介したK+の流出がひき起こされることによると考えられている15).リソソーム膜とNLRP3インフラマソームとの関係に関しては,リソソーム膜の損傷がNLRP3インフラマソームの活性化に関与することが報告されている13,16).リソソーム膜の破壊によりリソソームの内部に局在するタンパク質が細胞質に放出されることによりNLRP3インフラマソームは活性化されると考えられている.しかし,K+の流出およびリソソーム膜の損傷がどのような分子機構によりNLRP3インフラマソームを活性化するかについての詳細は明らかではない.ミトコンドリアの関与については,NLRP3インフラマソームが活性化するための足場を形成すること,ミトコンドリアに由来する分子がNLRP3インフラマソームの活性化に関与することが知られている.2012年,ミトコンドリアにより産生された活性酸素がミトコンドリアDNAを酸化させ,細胞質に移行してNLRP3と結合することが報告された17).ミトコンドリアを足場にするという点では,MVASがNLRP3と会合することがNLRP3インフラマソームの活性化において重要であることが報告された18).ただし,MVASを欠損した細胞ではATPに対する反応性が認められない一方,シリカなどの結晶に対する反応性は正常に誘導されることから,NLRP3インフラマソームの活性化のシグナル伝達経路として複数が存在しており,刺激により使い分けされていることが推測される.また,NLRP3インフラマソームの活性化には微小管を介した制御が必要であることも報告され19),NLRP3インフラマソームの活性化には特徴的な足場の形成が必要であることが明らかになった.
NLRP3遺伝子の変異に起因する疾患は遺伝型によりその症状が異なり,家族性感冒自己炎症性症候群,Muckle-Wells症候群,新生児期発症多臓器性炎症性疾患の3種類に大別される.常染色体優性の遺伝形式で発症する家族性感冒自己炎症性症候群およびMuckle-Wells症候群において,NLRP3遺伝子の活性化型の変異が同定された20).家族性感冒自己炎症性症候群は寒冷刺激による,かゆみをともなわないじんましん様の発疹,関節痛,発熱を特徴とする疾患である.寒冷刺激がどのような分子機構によりNLRP3インフラマソームを活性化するかについての詳細は理解されていない.Muckle-Wells症候群は発疹,関節痛,発熱にくわえ,感音性の難聴や腎アミロイドーシスを特徴とし,その発症には必ずしも寒冷刺激を必要としない.いずれもNLRP3遺伝子のヘテロな変異により発症することが明らかになっており,わが国でもその家系が報告されている21).つまり,NLRP3遺伝子の変異のパターンに依存して,軽症型である家族性感冒自己炎症性症候群から重症型であるMuckle-Wells症候群への一連の症状がひき起こされると考えられる.さらに,新生児期発症多臓器性炎症性疾患はより重症の疾患である.これらの病態は,NLRP3遺伝子の活性化型の変異によりカスパーゼ1が活性化しインターロイキン1βが過剰に産生されることに依存すると考えられており,事実,抗インターロイキン1β療法により顕著の効果が得られることが報告されている.
NLRP3遺伝子に生殖細胞変異の認められない新生児期発症多臓器性炎症性疾患の約70%がNLRP3遺伝子の変異を体細胞モザイクで発症するという注目すべき知見が報告された22).また,Muckle-Wells症候群においても同様の体細胞モザイクが報告された23).NLRP3遺伝子の変異をもつ細胞の数が全体の細胞に対し低い割合であっても,過剰なインターロイキン1βが産生されることが病態の本態であるため発症しうることが考えられる.
痛風は血液中の尿酸が高値になり,関節の腫脹や激しい痛みをともなう疾患である.痛風発作として知られる突然の激しい関節の痛みの特徴は,関節腔への好中球の流入である.以前から,尿酸一ナトリウムの結晶のひき起こす炎症が痛風の病態の基盤であることは知られていたが,その原因となるシグナル伝達経路は不明であった.2006年,NLRP3シグナル伝達経路が尿酸一ナトリウムにより活性化され,その結果として痛風が発症することが報告された12).事実,NLRP3ノックアウトマウスでは尿酸一ナトリウムによるインターロイキン1βの産生の亢進は観察されなかったことから,尿酸一ナトリウムとNLRP3との関連性が推測された.しかし,尿酸一ナトリウムがどのような分子機構によりNLRP3インフラマソームを活性化するかについてはいまだに不明な点があり,尿酸一ナトリウムの認識の機構およびそれにひきつづくNLRP3インフラマソームの活性化の機構の解明は今後の課題といえる.
アルツハイマー病は思考能力や記憶などが障害をうける進行性かつ不可逆性の脳疾患である.60歳以降に発症することが多く,認知症の原因のひとつである.アルツハイマー病の患者の脳にはアミロイド斑とよばれる多数の異常な凝集体が沈着していることが知られている.アルツハイマー病の患者の脳組織においてカスパーゼ1の活性化が観察された4).さらに,家族性アルツハイマー病にみられる遺伝子変異をもつカスパーゼ1ノックアウトマウスあるいはNLRP3ノックアウトマウスは,空間記憶などの障害が減弱していた.また,NLRP3の欠損によりミクログリアの表現型が変化し,βアミロイドの沈着が抑制されることも明らかになった.以上の結果から,NLRP3インフラマソームはアルツハイマー病の進行に関与していることが推測された.NLRP3シグナル伝達経路の抑制はアルツハイマー病の治療に有効である可能性が示唆され,進行性の脳疾患の代表であるアルツハイマー病の治療法の開発に期待がもたれる.
高脂肪食などによる肥満がインスリン抵抗性を誘導することが知られているが,それがどのような分子機序によるのかは不明である.最近の研究において,NLRP3がインスリン抵抗性に関与することが報告された.膵島アミロイドポリペプチドという2型糖尿病の発症に付随して膵臓に沈着することが知られているアミロイドのオリゴマーが,NLRP3シグナル伝達経路を活性化してインターロイキン1βの産生を亢進させることが見い出された24).また,2型糖尿病の治療薬であるグリブリドは膵島アミロイドポリペプチドによるインターロイキン1βの産生を抑制することも明らかにされた.また,血漿にもっとも豊富に存在する飽和脂肪酸であるパルミチン酸がNLRP3インフラマソームを活性化することによりインスリン抵抗性がひき起こされることが見い出された25).パルミチン酸はリポ多糖により刺激されたマクロファージからのインターロイキン1βの産生を亢進させるが,NLRP3あるいはカスパーゼ1を欠損したマクロファージではそのような亢進は観察されなかった.さらに,NLRP3ノックアウトマウスに高脂肪食をあたえても耐糖能およびインスリン感受性は悪化しないことも示された.パルミチン酸がどのような分子機構によりNLRP3インフラマソームを活性化するのかについては,パルミチン酸とリポ多糖によりAMPキナーゼのリン酸化が低下し,それによりオートファジーが低下することが見い出された.さらに,パルミチン酸はミトコンドリアからの活性酸素種の産生を亢進させていた.つまり,パルミチン酸およびリポ多糖はAMPキナーゼの阻害を介してオートファジーを抑制し,ミトコンドリアからの活性酸素種の産生を亢進させることによりNLRP3インフラマソームを活性化していると考えられた.
60年以上まえ,わが国において,進行性の脂肪の萎縮および高熱を特徴とする疾患が報告され,中条-西村症候群とよばれた.これらの疾患は古くからその存在が知られていたものの,その原因は不明であった.筆者らは,新潟および秋田に同様の疾患が存在することを知り,その患者は自己抗体をもたないこと,易感染性を示さないこと,激しい炎症が持続すること,という点で自己炎症性疾患に分類されうると考えた.患者の特徴的な臨床症状および検査所見として,高熱,結節性紅斑,高γグロブリン血症,血清C反応性タンパク質の高値にくわえ,著明な進行性の脂肪の萎縮や筋の萎縮などがあげられた.筆者らは,この疾患をJapanese autoinflammatory syndrome with lipodystrophy(JASL)と命名したが9),類似の症状を呈する疾患として,中条-西村症候群のほか,海外においてもJMP症候群およびCANDLE症候群が知られている8-11).
日本人の2つの家系を用いた連鎖解析およびホモ接合体マッピングにより,JASLの候補遺伝子は第6染色体に存在すると考えられた.その領域に焦点をあてたエクソーム解析の結果,原因となる遺伝子変異をPSMB8遺伝子のエキソン5に同定した9).この変異はアミノ酸置換をともなうミスセンス変異であった.このアミノ酸残基は種をこえて保存されていたことから,機能および構造の維持に重要であると推測された.PSMB8遺伝子はインターフェロンγにより発現の誘導されるプロテアソームの構成タンパク質であるβ5iをコードする遺伝子として報告されていた26).中条-西村症候群,JMP症候群,CANDLE症候群のいずれもがPSMB8遺伝子の変異により発症することも報告され8-11),わが国における変異はいずれも同じものであったが,海外で報告された変異は異なる部位の変異であった.一方,同様の症状を呈するがPSMB8遺伝子に変異の存在しない症例の存在することも明らかにされている.
プロテアソームはユビキチン化をうけたタンパク質を分解するタンパク分解酵素複合体である.プロテアソームはαリングにタンパク質分解酵素の活性をもつβサブユニットが結合するという基本構造をもち,現在までに,構成的に発現する標準プロテアソームのほか,免疫プロテアソームおよび胸腺プロテアソームが知られている27,28).標準プロテアソームはほとんどすべての細胞に発現し,構成タンパク質であるβ1,β2,β5がタンパク質分解酵素活性をもつ.標準プロテアソームのβ1,β2,β5がβ1i,β2i,β5iに置換したのが免疫プロテアソームである.標準プロテアソームと同様に,β1i,β2i,β5iがタンパク分解酵素活性をもち,そのうちβ5iは強いキモトリプシン様の活性をもつことが知られている.さきに述べたように,PSMB8遺伝子はこのβ5iをコードする.免疫プロテアソームはMHCクラスIに提示されるペプチドを効率よく切り出す役割をもつことが報告されている27).一方で,β5iノックアウトマウス,および,β1i,β2i,β5iのトリプルノックアウトマウスにおいて炎症は観察されていない29).この知見は,ヒトとマウスにおいて免疫プロテアソームの制御する機構が異なること,あるいは,それ以外の未知の機構のあることを示唆する.いずれの場合も,免疫プロテアソームの遺伝子変異に起因する自己炎症性疾患の存在は,恒常性の維持における免疫プロテアソームの機能的な役割を明らかにするうえできわめて貴重な知見であると考えられる(プロテアソームについての詳細は,村田 茂穂, 領域融合レビュー, 3, e011, 2014 を参照されたい).
JASLにおいて発見されたPSMB8遺伝子の変異はミスセンス変異であったが,この変異が免疫プロテアソームの機能の異常をきたすことについて,患者から樹立した不死化B細胞においてβ5iの発現が著明に低下していることが明らかになった.また,JASLに由来する不死化B細胞においては,β5iが主として制御するキモトリプシン様の活性だけではなく,トリプシン様の活性およびカスパーゼ様の活性のいずれもが低下しており,すべてのタンパク質分解酵素活性の低下していることが明らかになった.それでは,どのような分子機構によりすべてのタンパク質分解酵素活性が低下するのであろうか.プロテアソームにおいてはおのおののサブユニットによる複合体の形成が非常に厳密に制御されている.そこで,変異型のβ5iによりプロテアソームの分子集合が変化するかどうかについて検討した.その結果,変異型のβ5iの存在により免疫プロテアソームの分子集合は阻害され,β5iを含まない分子集合中間体が増加することが明らかになった.以上の結果から,PSMB8遺伝子のミスセンス変異により免疫プロテアソームの分子集合が阻害されることが推測された.実際に,JASLの患者の皮膚組織では細胞質および核においてユビキチン化タンパク質の蓄積が観察された.
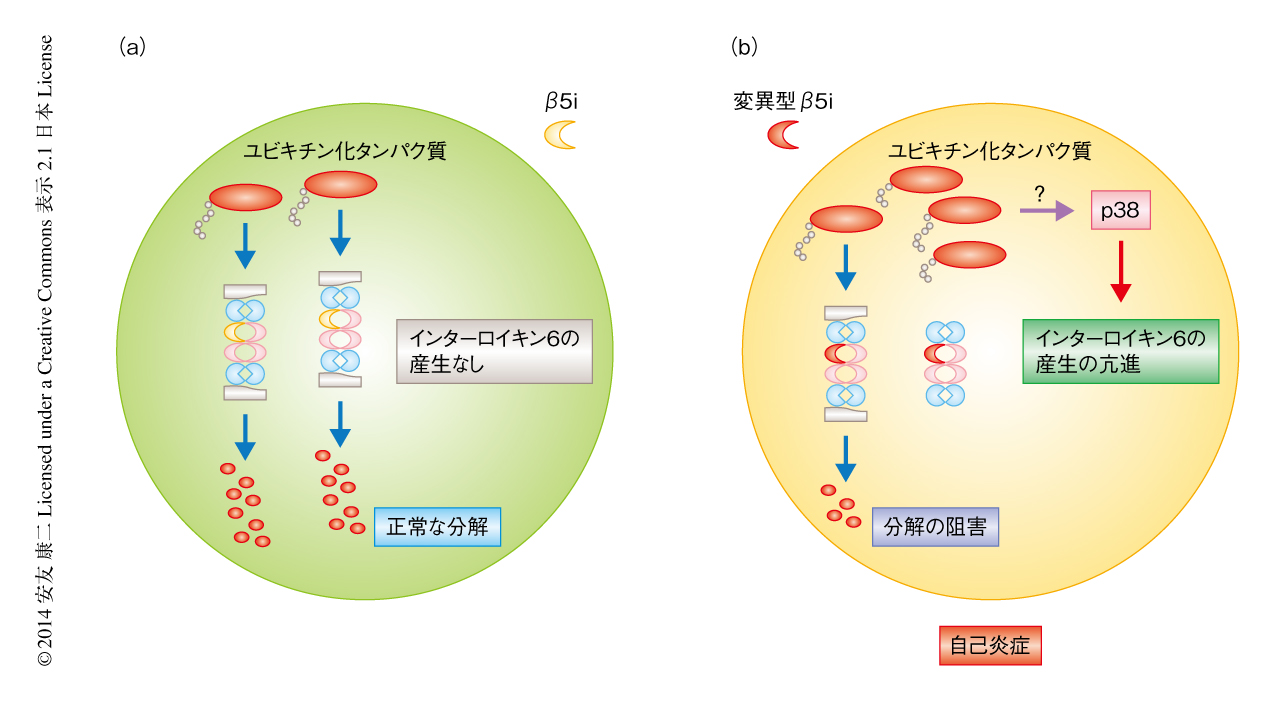
PSMB8ノックアウトマウスには炎症の自然発症は報告されていないことから,炎症がどのような機序により誘導されているかという点が重要な課題である.JASLの患者の皮膚においてはインターロイキン6の発現が上昇しており,JASLの患者に由来する不死化B細胞をPMAおよびイオノマイシンにより刺激すると,対照となる細胞に比べインターロイキン6の発現が有意に上昇した.また,このインターロイキン6の発現の上昇はp38阻害剤により抑制された.以上のことから,免疫プロテアソームの機能が低下することにより,過剰なp38を介してインターロイキン6の発現が増強していると考えられた(図2).しかしこれまでのところ,免疫プロテアソームの機能の異常がどのような分子機構によりp38を活性化させるかについては明らかにされていない.一方,インターフェロンγシグナル伝達経路が活性化しているという報告もあることから,複数のシグナル伝達経路がはたらくことにより炎症が誘導されていると考えられる.免疫プロテアソームの機能の低下により,どのような分子機序により各種のサイトカインの産生が増加し炎症が誘導されるかについては,JASLの動物モデルを樹立することなどにより明らかにする必要があると思われる.
JASLの特徴的な症状のひとつは進行性の脂肪の萎縮である.筆者らは,β5iの機能の低下が脂肪細胞の分化を抑制することが,脂肪の萎縮の原因のひとつであることを報告した9).JASLにおける脂肪の萎縮は免疫抑制剤などにより自己炎症が改善しても進行することから,臨床的には非常に重要な問題になる.脂肪細胞の分化の異常だけが脂肪の萎縮に関与しているのか,あるいは,炎症が臨床的に改善しても脂肪の萎縮は進行することから炎症の関与は低いとも考えられるが,炎症がどのくらい脂肪の萎縮に関与しているかについて明らかにする必要がある.
自己炎症性疾患として分類される疾患のほかにも,自己炎症がその進展にかかわる疾患は多様であると考えられる.遺伝性の自己炎症性疾患の研究から,自己炎症にかかわる遺伝子としてNOD様受容体ファミリーにくわえ,免疫プロテアソームの構成タンパク質が明らかにされた.おそらく,それ以外にも多様なシグナル伝達経路が自己炎症性疾患に寄与していると考えられ,今後,さらに遺伝学的な解析により原因となる遺伝的な素因に関する研究が進むと考えられる.また,そのシグナル伝達経路を遮断するような治療法の開発が,今後の炎症性疾患の治療法の開発の中心的な課題のひとつになると予測される.
略歴:1997年 徳島大学大学院医学研究科 修了,同年 米国NIH National Institute of Allergy and Infectious Diseases研究員,2000年 徳島大学医学部 医員,2011年 同 助手を経て,同年 同 教授(現 徳島大学大学院ヘルスバイオサイエンス研究部).
研究テーマ:ヒトの免疫疾患のゲノム解析,T細胞による免疫制御機構.
抱負:遺伝性免疫疾患の解析をつうじて,ヒトの免疫系における恒常性維持の機構および破綻の機序を理解したいと考えています.
研究室URL:http://immunology.hosp.med.tokushima-u.ac.jp/immunology/system/top/
© 2014 安友 康二 Licensed under CC 表示 2.1 日本
(徳島大学大学院ヘルスバイオサイエンス研究部 生体防御医学分野)
email:安友康二
領域融合レビュー, 3, e013 (2014) DOI: 10.7875/leading.author.3.e013
Koji Yasutomo: Molecular basis of autoinflammatory disorders.
要 約
自己抗原に応答するリンパ球がその発症の鍵となる自己免疫疾患が古くから免疫難病として知られている.それと対比される疾患として,自己炎症性疾患とよばれる疾患が家族性の炎症性疾患における原因遺伝子の研究から確立されてきた.自己炎症とは,自己免疫応答や明らかな感染が存在しないにもかかわらず炎症が誘導される病態であり,非常に幅広い疾患の基盤になると考えられる.最近のゲノム解析手法の発展により,自己炎症性の病態を誘導する遺伝子について解明が進みつつあり,NLRP3に代表されるNOD様受容体ファミリーの遺伝子異常にくわえ,最近,免疫プロテアソームの構成タンパク質の遺伝子異常による自己炎症性疾患が報告された.これらの遺伝子が単一因子性の疾患にとどまらず多因子性の疾患にも関与することも報告されつつあり,今後,自己炎症に関与する疾患はますます明らかになると予測される.それと並行して,自己炎症性の病態に焦点をあてた治療法の開発が今後の研究の中心的な課題になると思われる.
はじめに
炎症という病態の概念は非常に広範囲であり,炎症は古くから多くの疾患に寄与していると考えられてきた.一方で,炎症のかかわる疾患は多岐にわたるにもかかわらず,その分子機序の詳細はながらく明らかにされていなかった.ところが,家族性の炎症性疾患における原因遺伝子の発見をひとつの契機として1,2),炎症を誘導する分子機構の詳細がひもとかれつつある3).また同時に,悪性腫瘍や神経変性疾患などのように,これまで炎症との関連性が明確ではなかった疾患と炎症との関連性についても報告されるようになってきた4).以上のような背景から,炎症はこれまで考えられてきた以上に幅広い疾患の基盤となり,病態の進展に関与していると考えられるようになってきた.その考えにもとづき,現代の医療においては,どのように炎症を制御するかということが治療を考えるうえで中心的な課題になろうとしている.
炎症は内因性の異常,感染,毒物など外因性の物質などにより誘導されると考えられ,刺激に依存して発動される細胞内シグナル伝達経路やひき起こされる炎症の病態も多様である.免疫系の異常により誘導される疾患として古くから自己免疫疾患の概念が確立されており,自己免疫疾患は自己抗原を認識するT細胞およびB細胞が過剰に活性化あるいは増殖することにより発症すると考えられている.一方,最近の研究から,自己免疫応答や明らかな感染が存在しないにもかかわらず,自然免疫系を制御する細胞の過剰な活性化を契機とする激しい炎症を特徴とする疾患が見い出され,それらは自己炎症性疾患と定義されその概念が確立されつつある1,5).自己炎症性疾患は,狭義には単一の遺伝子の異常により発症する遺伝性の疾患をさすことが多いが,遺伝性が明らかではない疾患,あるいは,多因子性に発症すると考えられている疾患のなかにも自己炎症性疾患として分類の可能な疾患があると思われる.さらに,自己炎症性疾患を誘導するシグナル伝達経路はその進展の過程に寄与していると考えられることから,自己炎症は多岐にわたる疾患に寄与しているといえる(図1).
自己炎症性疾患の研究の進展は,NLRP3などの単一の因子の異常に起因する疾患の分子病態の発見が契機となり発展してきた1,2,6-11).そのような単一の遺伝子変異による疾患に関する研究から派生して,最近,NLRP3は糖尿病あるいはアルツハイマー病の病態にも寄与することが明らかになりつつある.このレビューでは,自己炎症性疾患の発症に寄与する分子の詳細とその病態について解説する.
1.NLRP3遺伝子の変異に起因する自己炎症性疾患
インフラマソームは細胞質に侵入した病原性微生物あるいは尿酸などの結晶により活性化するタンパク質複合体の総称である.一般的には,インフラマソームの活性化によりカスパーゼ1前駆体が活性をもつカスパーゼ1に変換し,インターロイキン1βやインターロイキン18などの炎症性サイトカインの産生を誘導することにより炎症を惹起することが知られている5).インフラマソームを構成するタンパク質としてはNOD様受容体が知られている.NOD様受容体ファミリーは細胞に感染する微生物を認識するタンパク質として進化の過程において多様性を獲得してきたと推測される.ここでは,NOD様受容体ファミリーのうち,自己炎症性疾患との関連性が深いNLRP3について解説する.
NLRP3インフラマソームはβアミロイド,尿酸の結晶,細胞外のATPにより活性化されることが知られている12,13).しかし,それらの分子がどのような分子機序によりNLRP3インフラマソームを活性化するかについての詳細はいまだ理解されていない.しかし,K+の流出やリソソーム膜の損傷などの機序が考えられており,さらに最近では,ミトコンドリアの関与などいくつかの重要な知見が報告されている.K+の関与に関しては,K+が細胞外に流出することにより細胞内のK+濃度が低下しNLRP3インフラマソームが活性化することが示されている14).実際に,K+の流出を抑制するとNLRP3インフラマソームの活性化が抑制されることも明らかにされている.高濃度のATPはNLRP3インフラマソームを活性化させるが,これはATPによりP2X7受容体を介したK+の流出がひき起こされることによると考えられている15).リソソーム膜とNLRP3インフラマソームとの関係に関しては,リソソーム膜の損傷がNLRP3インフラマソームの活性化に関与することが報告されている13,16).リソソーム膜の破壊によりリソソームの内部に局在するタンパク質が細胞質に放出されることによりNLRP3インフラマソームは活性化されると考えられている.しかし,K+の流出およびリソソーム膜の損傷がどのような分子機構によりNLRP3インフラマソームを活性化するかについての詳細は明らかではない.ミトコンドリアの関与については,NLRP3インフラマソームが活性化するための足場を形成すること,ミトコンドリアに由来する分子がNLRP3インフラマソームの活性化に関与することが知られている.2012年,ミトコンドリアにより産生された活性酸素がミトコンドリアDNAを酸化させ,細胞質に移行してNLRP3と結合することが報告された17).ミトコンドリアを足場にするという点では,MVASがNLRP3と会合することがNLRP3インフラマソームの活性化において重要であることが報告された18).ただし,MVASを欠損した細胞ではATPに対する反応性が認められない一方,シリカなどの結晶に対する反応性は正常に誘導されることから,NLRP3インフラマソームの活性化のシグナル伝達経路として複数が存在しており,刺激により使い分けされていることが推測される.また,NLRP3インフラマソームの活性化には微小管を介した制御が必要であることも報告され19),NLRP3インフラマソームの活性化には特徴的な足場の形成が必要であることが明らかになった.
NLRP3遺伝子の変異に起因する疾患は遺伝型によりその症状が異なり,家族性感冒自己炎症性症候群,Muckle-Wells症候群,新生児期発症多臓器性炎症性疾患の3種類に大別される.常染色体優性の遺伝形式で発症する家族性感冒自己炎症性症候群およびMuckle-Wells症候群において,NLRP3遺伝子の活性化型の変異が同定された20).家族性感冒自己炎症性症候群は寒冷刺激による,かゆみをともなわないじんましん様の発疹,関節痛,発熱を特徴とする疾患である.寒冷刺激がどのような分子機構によりNLRP3インフラマソームを活性化するかについての詳細は理解されていない.Muckle-Wells症候群は発疹,関節痛,発熱にくわえ,感音性の難聴や腎アミロイドーシスを特徴とし,その発症には必ずしも寒冷刺激を必要としない.いずれもNLRP3遺伝子のヘテロな変異により発症することが明らかになっており,わが国でもその家系が報告されている21).つまり,NLRP3遺伝子の変異のパターンに依存して,軽症型である家族性感冒自己炎症性症候群から重症型であるMuckle-Wells症候群への一連の症状がひき起こされると考えられる.さらに,新生児期発症多臓器性炎症性疾患はより重症の疾患である.これらの病態は,NLRP3遺伝子の活性化型の変異によりカスパーゼ1が活性化しインターロイキン1βが過剰に産生されることに依存すると考えられており,事実,抗インターロイキン1β療法により顕著の効果が得られることが報告されている.
NLRP3遺伝子に生殖細胞変異の認められない新生児期発症多臓器性炎症性疾患の約70%がNLRP3遺伝子の変異を体細胞モザイクで発症するという注目すべき知見が報告された22).また,Muckle-Wells症候群においても同様の体細胞モザイクが報告された23).NLRP3遺伝子の変異をもつ細胞の数が全体の細胞に対し低い割合であっても,過剰なインターロイキン1βが産生されることが病態の本態であるため発症しうることが考えられる.
痛風は血液中の尿酸が高値になり,関節の腫脹や激しい痛みをともなう疾患である.痛風発作として知られる突然の激しい関節の痛みの特徴は,関節腔への好中球の流入である.以前から,尿酸一ナトリウムの結晶のひき起こす炎症が痛風の病態の基盤であることは知られていたが,その原因となるシグナル伝達経路は不明であった.2006年,NLRP3シグナル伝達経路が尿酸一ナトリウムにより活性化され,その結果として痛風が発症することが報告された12).事実,NLRP3ノックアウトマウスでは尿酸一ナトリウムによるインターロイキン1βの産生の亢進は観察されなかったことから,尿酸一ナトリウムとNLRP3との関連性が推測された.しかし,尿酸一ナトリウムがどのような分子機構によりNLRP3インフラマソームを活性化するかについてはいまだに不明な点があり,尿酸一ナトリウムの認識の機構およびそれにひきつづくNLRP3インフラマソームの活性化の機構の解明は今後の課題といえる.
アルツハイマー病は思考能力や記憶などが障害をうける進行性かつ不可逆性の脳疾患である.60歳以降に発症することが多く,認知症の原因のひとつである.アルツハイマー病の患者の脳にはアミロイド斑とよばれる多数の異常な凝集体が沈着していることが知られている.アルツハイマー病の患者の脳組織においてカスパーゼ1の活性化が観察された4).さらに,家族性アルツハイマー病にみられる遺伝子変異をもつカスパーゼ1ノックアウトマウスあるいはNLRP3ノックアウトマウスは,空間記憶などの障害が減弱していた.また,NLRP3の欠損によりミクログリアの表現型が変化し,βアミロイドの沈着が抑制されることも明らかになった.以上の結果から,NLRP3インフラマソームはアルツハイマー病の進行に関与していることが推測された.NLRP3シグナル伝達経路の抑制はアルツハイマー病の治療に有効である可能性が示唆され,進行性の脳疾患の代表であるアルツハイマー病の治療法の開発に期待がもたれる.
高脂肪食などによる肥満がインスリン抵抗性を誘導することが知られているが,それがどのような分子機序によるのかは不明である.最近の研究において,NLRP3がインスリン抵抗性に関与することが報告された.膵島アミロイドポリペプチドという2型糖尿病の発症に付随して膵臓に沈着することが知られているアミロイドのオリゴマーが,NLRP3シグナル伝達経路を活性化してインターロイキン1βの産生を亢進させることが見い出された24).また,2型糖尿病の治療薬であるグリブリドは膵島アミロイドポリペプチドによるインターロイキン1βの産生を抑制することも明らかにされた.また,血漿にもっとも豊富に存在する飽和脂肪酸であるパルミチン酸がNLRP3インフラマソームを活性化することによりインスリン抵抗性がひき起こされることが見い出された25).パルミチン酸はリポ多糖により刺激されたマクロファージからのインターロイキン1βの産生を亢進させるが,NLRP3あるいはカスパーゼ1を欠損したマクロファージではそのような亢進は観察されなかった.さらに,NLRP3ノックアウトマウスに高脂肪食をあたえても耐糖能およびインスリン感受性は悪化しないことも示された.パルミチン酸がどのような分子機構によりNLRP3インフラマソームを活性化するのかについては,パルミチン酸とリポ多糖によりAMPキナーゼのリン酸化が低下し,それによりオートファジーが低下することが見い出された.さらに,パルミチン酸はミトコンドリアからの活性酸素種の産生を亢進させていた.つまり,パルミチン酸およびリポ多糖はAMPキナーゼの阻害を介してオートファジーを抑制し,ミトコンドリアからの活性酸素種の産生を亢進させることによりNLRP3インフラマソームを活性化していると考えられた.
2.免疫プロテアソームの機能の不全に起因する自己炎症性疾患
60年以上まえ,わが国において,進行性の脂肪の萎縮および高熱を特徴とする疾患が報告され,中条-西村症候群とよばれた.これらの疾患は古くからその存在が知られていたものの,その原因は不明であった.筆者らは,新潟および秋田に同様の疾患が存在することを知り,その患者は自己抗体をもたないこと,易感染性を示さないこと,激しい炎症が持続すること,という点で自己炎症性疾患に分類されうると考えた.患者の特徴的な臨床症状および検査所見として,高熱,結節性紅斑,高γグロブリン血症,血清C反応性タンパク質の高値にくわえ,著明な進行性の脂肪の萎縮や筋の萎縮などがあげられた.筆者らは,この疾患をJapanese autoinflammatory syndrome with lipodystrophy(JASL)と命名したが9),類似の症状を呈する疾患として,中条-西村症候群のほか,海外においてもJMP症候群およびCANDLE症候群が知られている8-11).
日本人の2つの家系を用いた連鎖解析およびホモ接合体マッピングにより,JASLの候補遺伝子は第6染色体に存在すると考えられた.その領域に焦点をあてたエクソーム解析の結果,原因となる遺伝子変異をPSMB8遺伝子のエキソン5に同定した9).この変異はアミノ酸置換をともなうミスセンス変異であった.このアミノ酸残基は種をこえて保存されていたことから,機能および構造の維持に重要であると推測された.PSMB8遺伝子はインターフェロンγにより発現の誘導されるプロテアソームの構成タンパク質であるβ5iをコードする遺伝子として報告されていた26).中条-西村症候群,JMP症候群,CANDLE症候群のいずれもがPSMB8遺伝子の変異により発症することも報告され8-11),わが国における変異はいずれも同じものであったが,海外で報告された変異は異なる部位の変異であった.一方,同様の症状を呈するがPSMB8遺伝子に変異の存在しない症例の存在することも明らかにされている.
プロテアソームはユビキチン化をうけたタンパク質を分解するタンパク分解酵素複合体である.プロテアソームはαリングにタンパク質分解酵素の活性をもつβサブユニットが結合するという基本構造をもち,現在までに,構成的に発現する標準プロテアソームのほか,免疫プロテアソームおよび胸腺プロテアソームが知られている27,28).標準プロテアソームはほとんどすべての細胞に発現し,構成タンパク質であるβ1,β2,β5がタンパク質分解酵素活性をもつ.標準プロテアソームのβ1,β2,β5がβ1i,β2i,β5iに置換したのが免疫プロテアソームである.標準プロテアソームと同様に,β1i,β2i,β5iがタンパク分解酵素活性をもち,そのうちβ5iは強いキモトリプシン様の活性をもつことが知られている.さきに述べたように,PSMB8遺伝子はこのβ5iをコードする.免疫プロテアソームはMHCクラスIに提示されるペプチドを効率よく切り出す役割をもつことが報告されている27).一方で,β5iノックアウトマウス,および,β1i,β2i,β5iのトリプルノックアウトマウスにおいて炎症は観察されていない29).この知見は,ヒトとマウスにおいて免疫プロテアソームの制御する機構が異なること,あるいは,それ以外の未知の機構のあることを示唆する.いずれの場合も,免疫プロテアソームの遺伝子変異に起因する自己炎症性疾患の存在は,恒常性の維持における免疫プロテアソームの機能的な役割を明らかにするうえできわめて貴重な知見であると考えられる(プロテアソームについての詳細は,村田 茂穂, 領域融合レビュー, 3, e011, 2014 を参照されたい).
JASLにおいて発見されたPSMB8遺伝子の変異はミスセンス変異であったが,この変異が免疫プロテアソームの機能の異常をきたすことについて,患者から樹立した不死化B細胞においてβ5iの発現が著明に低下していることが明らかになった.また,JASLに由来する不死化B細胞においては,β5iが主として制御するキモトリプシン様の活性だけではなく,トリプシン様の活性およびカスパーゼ様の活性のいずれもが低下しており,すべてのタンパク質分解酵素活性の低下していることが明らかになった.それでは,どのような分子機構によりすべてのタンパク質分解酵素活性が低下するのであろうか.プロテアソームにおいてはおのおののサブユニットによる複合体の形成が非常に厳密に制御されている.そこで,変異型のβ5iによりプロテアソームの分子集合が変化するかどうかについて検討した.その結果,変異型のβ5iの存在により免疫プロテアソームの分子集合は阻害され,β5iを含まない分子集合中間体が増加することが明らかになった.以上の結果から,PSMB8遺伝子のミスセンス変異により免疫プロテアソームの分子集合が阻害されることが推測された.実際に,JASLの患者の皮膚組織では細胞質および核においてユビキチン化タンパク質の蓄積が観察された.
PSMB8ノックアウトマウスには炎症の自然発症は報告されていないことから,炎症がどのような機序により誘導されているかという点が重要な課題である.JASLの患者の皮膚においてはインターロイキン6の発現が上昇しており,JASLの患者に由来する不死化B細胞をPMAおよびイオノマイシンにより刺激すると,対照となる細胞に比べインターロイキン6の発現が有意に上昇した.また,このインターロイキン6の発現の上昇はp38阻害剤により抑制された.以上のことから,免疫プロテアソームの機能が低下することにより,過剰なp38を介してインターロイキン6の発現が増強していると考えられた(図2).しかしこれまでのところ,免疫プロテアソームの機能の異常がどのような分子機構によりp38を活性化させるかについては明らかにされていない.一方,インターフェロンγシグナル伝達経路が活性化しているという報告もあることから,複数のシグナル伝達経路がはたらくことにより炎症が誘導されていると考えられる.免疫プロテアソームの機能の低下により,どのような分子機序により各種のサイトカインの産生が増加し炎症が誘導されるかについては,JASLの動物モデルを樹立することなどにより明らかにする必要があると思われる.
JASLの特徴的な症状のひとつは進行性の脂肪の萎縮である.筆者らは,β5iの機能の低下が脂肪細胞の分化を抑制することが,脂肪の萎縮の原因のひとつであることを報告した9).JASLにおける脂肪の萎縮は免疫抑制剤などにより自己炎症が改善しても進行することから,臨床的には非常に重要な問題になる.脂肪細胞の分化の異常だけが脂肪の萎縮に関与しているのか,あるいは,炎症が臨床的に改善しても脂肪の萎縮は進行することから炎症の関与は低いとも考えられるが,炎症がどのくらい脂肪の萎縮に関与しているかについて明らかにする必要がある.
おわりに
自己炎症性疾患として分類される疾患のほかにも,自己炎症がその進展にかかわる疾患は多様であると考えられる.遺伝性の自己炎症性疾患の研究から,自己炎症にかかわる遺伝子としてNOD様受容体ファミリーにくわえ,免疫プロテアソームの構成タンパク質が明らかにされた.おそらく,それ以外にも多様なシグナル伝達経路が自己炎症性疾患に寄与していると考えられ,今後,さらに遺伝学的な解析により原因となる遺伝的な素因に関する研究が進むと考えられる.また,そのシグナル伝達経路を遮断するような治療法の開発が,今後の炎症性疾患の治療法の開発の中心的な課題のひとつになると予測される.
文 献
- McDermott, M. F., Aksentijevich, I., Galon, J. et al.: Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell, 97, 133-144 (1999)[PubMed]
- Park, H., Bourla, A. B., Kastner, D. L. et al.: Lighting the fires within: the cell biology of autoinflammatory diseases. Nat. Rev. Immunol., 12, 570-580 (2012)[PubMed]
- Ombrello, M. J. & Kastner, D. L.: Autoinflammation in 2010: expanding clinical spectrum and broadening therapeutic horizons. Nat. Rev. Rheumatol., 7, 82-84 (2011)[PubMed]
- Heneka, M. T., Kummer, M. P., Stutz, A. et al.: NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature, 493, 674-678 (2013)[PubMed]
- Kastner, D. L., Aksentijevich, I. & Goldbach-Mansky, R.: Autoinflammatory disease reloaded: a clinical perspective. Cell, 140, 784-790 (2010)[PubMed]
- French FMF Consortium: A candidate gene for familial Mediterranean fever. Nat. Genet., 17, 25-31 (1997)[PubMed]
- Drenth, J. P., Cuisset, L., Grateau, G. et al.: Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat. Genet., 22, 178-181 (1999)[PubMed]
- Agarwal, A. K., Xing, C., DeMartino, G. N. et al.: PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am. J. Hum. Genet., 87, 866-872 (2010)[PubMed]
- Kitamura, A., Maekawa, Y., Uehara, H. et al.: A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Invest., 121, 4150-4160 (2011)[PubMed]
- Arima, K., Kinoshita, A., Mishima, H. et al.: Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc. Natl. Acad. Sci. USA, 108, 14914-14919 (2011)[PubMed]
- Liu, Y., Ramot, Y., Torrelo, A. et al.: Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Heum., 64, 895-907 (2012)[PubMed]
- Martinon, F., Petrilli, V., Mayor, A. et al.: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature, 440, 237-241 (2006)[PubMed]
- Halle, A., Hornung, V., Petzold, G. C. et al.: The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol., 9, 857-865 (2008)[PubMed]
- Petrilli, V., Papin, S., Dostert, C. et al.: Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ., 14, 1583-1589 (2007)[PubMed]
- Lamkanfi, M., Mueller, J. L., Vitari, A. C. et al.: Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol., 187, 61-70 (2009)[PubMed]
- Hornung, V., Bauernfeind, F., Halle, A. et al.: Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol., 9, 847-856 (2008)[PubMed]
- Shimada, K., Crother, T. R., Karlin, J. et al.: Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity, 36, 401-414 (2012)[PubMed]
- Subramanian, N., Natarajan, K., Clatworthy, M. R. et al.: The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell, 153, 348-361 (2013)[PubMed]
- Misawa, T., Takahama, M., Kozaki, T. et al.: Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol., 14, 454-460 (2013)[PubMed]
- Hoffman, H. M., Mueller, J. L., Broide, D. H. et al.: Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet., 29, 301-305 (2001)[PubMed]
- Kanegane, H., Itazawa, T. Saito, M. et al.: A CIAS1 mutation in a Japanese girl with familial cold autoinflammatory syndrome. Eur. J. Pediatr., 167, 245-247 (2008)[PubMed]
- Saito, M., Fujisawa, A., Nishikomori, R. et al.: Somatic mosaicism of CIAS1 in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum., 52, 3579-3585 (2005)[PubMed]
- Tanaka, N., Izawa, K., Saito, M. K. et al.: High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum., 63, 3625-3632 (2011)[PubMed]
- Masters, S. L., Dunne, A., Subramanian, S. L. et al.: Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol., 11, 897-904 (2010)[PubMed]
- Wen, H., Gris, D., Lei, Y. et al.: Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol., 12, 408-415 (2011)[PubMed]
- Akiyama, K., Yokota, K., Kagawa, S. et al.: cDNA cloning and interferon gamma down-regulation of proteasomal subunits X and Y. Science, 265, 1231-1234 (1994)[PubMed]
- Murata, S., Yashiroda, H. & Tanaka, K.: Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol., 10, 104-115 (2009)[PubMed]
- Murata, S., Takahama, Y. & Tanaka, K.: Thymoproteasome: probable role in generating positively selecting peptides. Curr. Opin. Immunol., 20, 192-196 (2008)[PubMed]
- Kincaid, E. Z., Che, J. W., York, I. et al.: Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol., 13, 129-135 (2012)[PubMed]
著者プロフィール
略歴:1997年 徳島大学大学院医学研究科 修了,同年 米国NIH National Institute of Allergy and Infectious Diseases研究員,2000年 徳島大学医学部 医員,2011年 同 助手を経て,同年 同 教授(現 徳島大学大学院ヘルスバイオサイエンス研究部).
研究テーマ:ヒトの免疫疾患のゲノム解析,T細胞による免疫制御機構.
抱負:遺伝性免疫疾患の解析をつうじて,ヒトの免疫系における恒常性維持の機構および破綻の機序を理解したいと考えています.
研究室URL:http://immunology.hosp.med.tokushima-u.ac.jp/immunology/system/top/
© 2014 安友 康二 Licensed under CC 表示 2.1 日本
