次世代シークエンサーにより得られたデータの解析
2015/05/18
坊農 秀雅
(ライフサイエンス統合データベースセンター)
email:坊農秀雅
領域融合レビュー, 4, e008 (2015) DOI: 10.7875/leading.author.4.e008
Hidemasa Bono: Sequence data analysis in life science utilizing next generation sequencers.

生命科学の研究において次世代シークエンサーが普通に使われるようになってきた.これまで,さまざまな応用が提案されてきたが,最近では,ゲノムの再解読による多型の解析やゲノムの新規な解読,トランスクリプトームの解読によるRNA転写量の測定,DNA結合タンパク質の結合配列の解析,細菌叢のメタゲノムの解析がおもなものになった.対応するデータ解析の手法もほぼ固まってきたようにみえる.そこで,このレビューでは,次世代シークエンサーにより得られたデータの解析手法を,公共データベースのデータを解析してきた立場から紹介する.
次世代シークエンサー(next generation sequencer:NGS)により解読された塩基配列の情報は,どのような実験を行ったかというメタデータとともに,SRA(Sequence Read Archive)とよばれる公共データベースに登録されている1).次世代シークエンサーにより得られたデータの登録は2007年からはじまり,2015年4月現在,総塩基数で約3.6ペタ塩基(ペタは10の15乗),データ量は約2.3ペタバイトと,保持するだけでもたいへんな量になっている(http://www.ncbi.nlm.nih.gov/Traces/sra/).その研究分野による内訳をみると,ゲノムが3/4近くをしめ,その残りの半分がトランスクリプトーム,ついでメタゲノムなっている(図1).このレビューでは,次世代シークエンサーにより得られたデータの解析手法を解説する.

次世代シークエンサーにより得られたデータの解析に用いられるソフトウェアの多くはオープンソースで無償で使えるものであり,多くのユーザーがそれをテストし各種のメーリングリストやtwitterなどのソーシャルメディアでその評判が流布している.それらをまとめたSEQanswersのWikiには,2014年4月15日現在,690ものソフトウェアが登録されている(http://seqanswers.com/wiki/Software).数多くのソフトウェアが存在するものの,やっていること自体はほぼ同じというものが多く,ソフトウェアの種類自体はほぼ出尽くした感がある.それらのうち代表的なものを紹介する.これら次世代シークエンサーに関連する配列データのフォーマットをまとめた(表1).

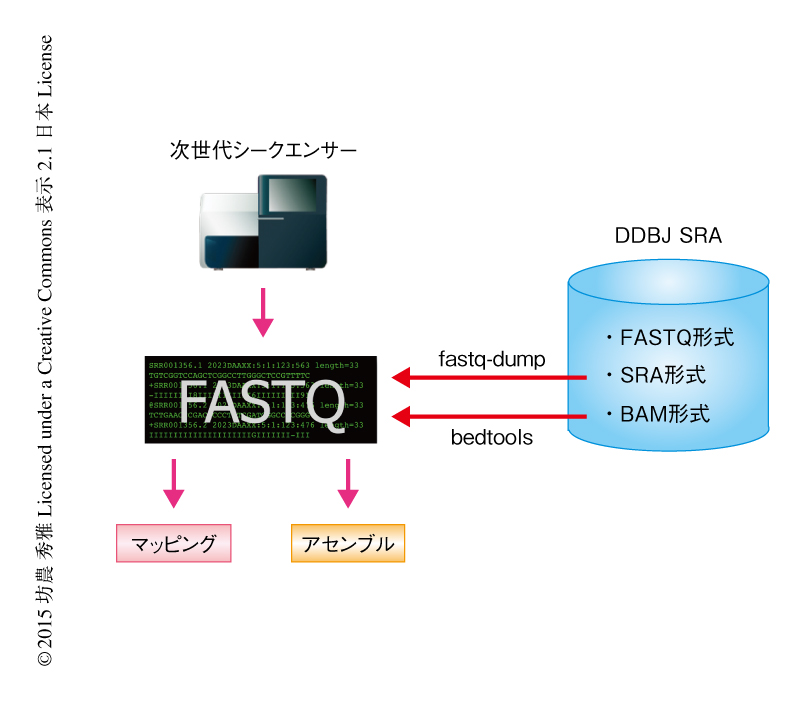
次世代シークエンサーから直接に得るにしても,SRAなどの公共データベースからダウンロードするにしても,データ解析のハブはFASTQ形式の配列ファイルである(図2).そのFASTQファイルをもとに,データを解析する前処理としてアダプター配列やタグ配列を除去し品質管理を行うが,その目的にはFASTQCというソフトウェアがよく用いられる(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).そののち,データ解析はリファレンスとなるゲノムにマッピングするか,アセンブルするかに分かれる.
リファレンス配列がすでにあるヒトや,ゲノム配列がすでに解読されているマウス,ショウジョウバエ,線虫などの多くの古典的なモデル生物においては,次世代シークエンサーにより得られたリード配列をリファレンスとなるゲノム配列に対して“貼りつけ”(マッピング)をすることからデータ解析がはじまる.このマッピングのためのソフトウェアとしては,Bowtie 2) あるいはBWA 3) が使われることがほとんどである.マッピングには大きな計算コストがかかり,多くの計算時間およびメモリを要する.その出力結果はゲノムに対するBAM形式のアラインメントファイルとして得られる.Bowtieの出力結果はBAM形式のテキスト版であるSAM形式により得られるが,そののちのデータ処理の入力がソートされたBMA形式であることが多いので,samtoolsというソフトウェアを用いてBAM形式に変換しソートすることが多い4).最新版のsamtools(version1.2)ではこのソートの並列化が実装され変換の高速化が図られている.
リファレンスとなるゲノム配列のない生物種ではマッピングはできないなので,次世代シークエンサーにより得られたリード配列の“つなぎあわせ”(アセンブル)をする必要がある.アセンブルにより得られるのは,BLASTなど配列類似性の検索でおなじみのFASTA形式の配列データである.
次世代シークエンサーがもっとも使われているのは,個体のあいだのゲノムの解析,とくに,ヒト個人のゲノム解読である.ヒトゲノム全体の1%ほどのmRNAに転写されるエキソン領域のみを再解読の対象とするエキソーム解析では,マッピングのためのソフトウェアとしてBWAを使う解析フローが紹介されることが多い.その一例を簡単に述べると,マッピングにより得られたBAMファイルから,samtoolsやPicard(http://picard.sourceforge.net/)を使い重複のあるリード配列を除き,Bedtoolsを用いてエキソン領域のみを抽出する5).そして,samtoolsにより多型(とくに1塩基置換,single nucleotide variant:SNV)のある場所を抽出し,非同義置換,ミスセンス変異,フレームシフト変異というアノテーションをつけ,VCF形式のファイルとして結果を得る.
また,エピゲノムを解析する場合には,バイサルファイト処理によりメチル化されなかったシトシンがウラシルに置換されDNA配列を解読する際にチミンとして読まれることを利用してメチル化された部位を見い出すWGBS(whole genome bisulfite sequencing)法により解析する.メチル化された部位に塩基置換が起こるのでそれを1塩基置換として見い出す戦略をとり,基本的なデータ解析の手法はゲノムの再解読のときと同じである.最終的には,ゲノムブラウザとしてよく使われるIGV 6) を用いて,候補となる領域を研究者が自分の目で確認する.
また,ゲノムを新規に解読する場合にはアセンブルの必要があるが,そのためのソフトウェア(アセンブラー)は多く開発されており,たとえば,nucleotid.esというWebサイト(http://nucleotid.es/)にカタログ化されている.なかでも,米国Broad Instituteにおいて開発されたALLPATH-LG 7) や,日本発のPlatanus 8) などがよく使われている.これらは無償で利用できるが,アセンブルのためのソフトウェアは一般に大きなメモリが必要となるので個人や研究室の所有するマシンでは動かせないことが多い.そこで,スーパーコンピューター(スパコン)を利用することになるが,研究目的なら国立遺伝学研究所のスパコンにおいてさまざまなソフトウェアが試用できるので利用するとよい.また,有償のソフトウェアとしては,デンマークのCLC bio社が開発しているCLC assembly cell(http://www.clcbio.com/products/clc-assembly-cell/)はスパコンにしかない大きなメモリを必要とせずMacOSXでも実行が可能で,さまざまな理由から外部に出せない配列データのアセンブルに適している.アセンブルによりコンティグが得られたら,それらの順序および向きをそろえてより長い配列を得る必要がある.それをやってくれるのがOperaというソフトウェアである9).まず,FASTA形式のコンティグのファイルとそれを生成するのに使ったリード配列のFASTQ形式のファイル,マッピングに使うソフトウェア(BWAあるいはBowtie)を引数にあたえて実行し,map形式の結果ファイルを得る.そののち,Operaを起動してFASTA形式の配列ファイルを得ることにより,より少なく,かつ,平均的により長くなったコンティグが得られる.得らえたゲノム配列は,近縁種のcDNAやアミノ酸配列に対する配列類似性検索によりアノテーションし,最終的にはGTF形式(GFF形式)のファイルを得る.
マイクロアレイを用いたハイブリダイゼーション法をベースにした手法が主流であったRNA転写量の測定も,次世代シークエンサーを用いたRNA-seq法がとって代わろうとしている.これは,転写されたRNAの配列をすべて解読し,それぞれの個数をそれが由来する転写単位(遺伝子)の発現強度とする手法である.かつて,Bodymap法とよばれる手法では,EST(expressed sequence tag)とよばれるmRNAの配列断片をクラスタリングし,転写単位ごとにその数を数えあげることにより遺伝子発現量を解析していた10).RNA-seq法は,まさに次世代シークエンサーを使うことによりこのBodymap法をなしとげるものである.RNA-seq法により得られるRNA転写量の単位として,RPKM(reads per kilobase per million mapped reads)がよく用いられる.これは正規化された遺伝子発現量で,100万個のリード配列をマッピングし転写産物の長さを1000塩基としたときのマッピングされたリード配列の数である11).また,RPKMの代わりに使われることの多いFPKM(fragments per kilobase of exon per million mapped)もほぼ同じで,断片ごとの正規化された遺伝子発現量である.RNA-seq法に関しても,リファレンスとなるゲノムにマッピングするかアセンブルするかに分かれる.
マッピングによる方法では,エキソーム解析と同じくBowtieやBWAというマッピングのためのソフトウェアが使われる.しかし,RNAに特有のスプライシングに対するアラインメントが必要になる.それを行うのがTopHatというソフトウェアで,TopHatが内部でBowtieを起動するため,RNA-seq法ではマッピングにBowtieが使われることが多い12).そののち,ゲノムのどの位置に遺伝子があるかなどを記述したゲノムアノテーションのファイル(多くの場合,GTF形式)を使い,Cufflinksというソフトウェアにより選択的スプライシングによるスプライスバリアントをリストアップする13).Cufflinksにより出力される遺伝子発現量はFPKMである.Cufflinksは複数のソフトウェアからなり,いくつかの計算ステップが必要ではあるが,Cuffdiffにより指定した2つの状態の遺伝子発現量の差を同定することができる.さらに,Cufflinksの結果を読み込んでR/Bioconductorにおいて便利に使えるようにするcummeRbundというパッケージもある.トランスクリプトーム解析のための多くのソフトウェアがR/Bioconductorで開発されていることもあり,さらなるデータ解析が進めやすく便利である14).ただし,TopHat(Bowtie)を実行したのちにCufflinksを実行するという一連の過程は必要なメモリの量が多く,多くのCPUが搭載されているマシンであっても1つのCPUで実行される部分もあるなど,計算には数時間のオーダーがかかる.そこで,より高速なソフトウェアの開発が進められ,最近では,アラインメントをせずにk-merをカウントすることにより遺伝子発現を定量する方法が注目されている.その代表的なソフトウェアにSailfishがある15).このソフトウェアはトランスクリプトームが既知でないと利用できないが,いちどインデックスを作成さえしておけば,あとはFASTQファイルごとにかなり高速に遺伝子発現が定量できる.
アセンブルによる方法では,Trinityというソフトウェアがよく使われる16).必要なメモリの量が多く計算時間も長くかかるのが難点であるが,国立遺伝学研究所のスパコンでも試用できる.
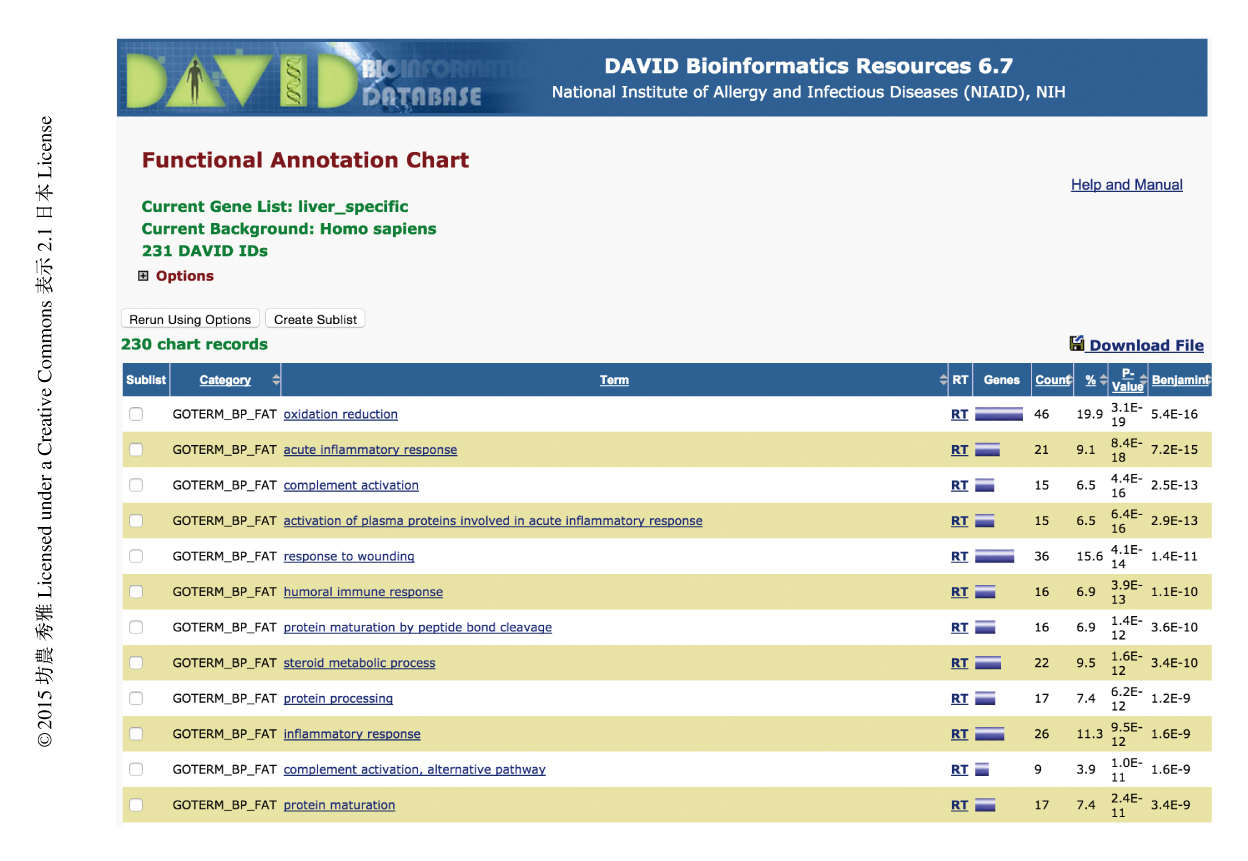
ここまでの手順により遺伝子単位で発現量を定量してしまえば,マイクロアレイ解析での手順がほぼそのまま利用できる.抽出した遺伝子セットが遺伝子全体からみてどういう特徴をもつのかみるのによく用いられるのがGSEA(gene set enrichment analysis)法である17).もちろん,エキソーム解析やのちに述べるChIP-seq法などにおいてもこのGSEA法は有効である.なかでも,DAVIDとよばれるウェブツールはインターフェースもよくできており便利である18).OMIM,Gene Ontology,Pathwayという機能情報への遺伝子アノテーションを利用したデータの解釈は,マイクロアレイにおけるデータ解析と同様に,次世代シークエンサーにより得られた大量のデータを解釈する手段として有効である(図3).さらに最近,論文データベースであるPubMedにおいておのおのの論文に付与されているMeSH(medical subject headings)を使いGSEA法により解析する手法を実装したR/Bioconductorのパッケージmeshrが公開され,データ解析のバリエーションがさらに広がった19).
以前より,DNA結合タンパク質の結合したDNA配列を解析する手法としてクロマチン免疫沈降(chromatin immunoprecipitation:ChIP)法があり,2000年代前半から2000年代中ごろにかけては,DNAの配列断片をマイクロアレイにより検出するChIP-on-chip法が用いられていた.そして,マイクロアレイの代わりに次世代シークエンサーを用いてDNAの配列断片を解読する方法が開発されChIP-seq法とよばれるようになった20).ChIP-seq法では,DNA結合タンパク質を認識する抗体を用いてこれが結合したDNAの配列断片を回収し解析する.ターゲットとなるタンパク質としてヒストンと転写因子がある.ともに結合したDNA配列を解読することによりゲノムのどの領域に結合していたかを知ることができ,ゲノムのどの位置に遺伝子があるかというゲノムアノテーション情報とつきあわせることにより直接的な転写制御の関係が推定できる.
ChIP-seq法においては,クロマチン免疫沈降法によりDNA結合タンパク質に結合したDNAの配列断片をリファレンスとなるゲノムにマッピングし,得られたBAM形式のアラインメントファイルを入力として,MACSというソフトウェアを使い結合部位を推定する21).得られる結果は,染色体の番号,その場所(startとend)とその場所でのピークの値がかかれたBED形式のファイルである.この情報からゲノムのどこにピークがあるかがわかる.そのゲノムにおける位置がどこか,ゲノムアノテーション情報をもとにR/Bioconductorなどを使い解析する.ChIP-seq法におけるデータ解析にあたりむずかしいのは,“結合がある”とするかどうかの閾値の線引きで,こればかりは実際のデータをみて個々に決めていく必要がある.また,得られた結合配列の特徴(転写因子の場合には,転写因子結合配列モチーフ)を知りたい場合には,それらの配列を抽出しアラインメントしたのち,その配列の特徴をWebLogoを用いて頻出する塩基が大きく表示されるよう可視化するなどの手段がある22).
次世代シークエンサーにより細菌叢の全体がもつDNAの配列をいちどに解読できるようになり,ショットガン法により細菌叢の全ゲノムを解析する方法や,16S rRNAのみを解読し細菌叢に存在する各種の細菌の割合を解析する方法が開発されている.その結果,ヒト腸内細菌叢のメタゲノムは,ヒト,マウスについで公共データベースSRAに多く登録されている.16S rRNAを解析する方法は,解読されたリード配列に含まれる16S rRNAの配列がデータベース化された既知の16S rRNAの配列のどれにマッチするかを探索するものである.全ゲノムの解析については,微生物における全ゲノム配列の解読と同じステップをふむ.まず,解読されたリード配列を既知のデータベースにたよることなくアセンブルする.その配列からタンパク質の配列をコードするORFを見い出し,得られたアミノ酸配列を質問配列として,これまでに知られているアミノ酸配列のデータベースに対しBLASTなどを使い配列類似性を検索する.その結果から,配列類似性にもとづき機能アノテーションし,最終的に,KEGGやCOGという分類のためのデータベースを用いて機能分類をする.
次世代シークエンサーにより得られたデータは基本的には公共データベースであるSRAに登録される.それは,解析結果の再現性の担保のため,そして,科学の進展のためであり,現在,われわれが思いもよらないような使い方がされそこから大発見があるかもしれないからである.例をあげるなら,かつて理化学研究所のFANTOMプロジェクトにおいて作製されたESTデータベースは,iPS細胞の分化を誘導する4つの因子を絞り込むことに使われた(http://www.osc.riken.jp/contents/fantom/).今後も同じようなことが起こるよう,次世代シークエンサーにより得られたデータは公共データベースへ登録していくべきなのである.わが国のDDBJ(DNA Data Bank of Japan)は米国のNCBIや欧州のEBIとデータを交換しているので,DDBJのDRA(DDBJ Sequence Read Archive)に登録してもSRAに登録するのと同じであり,大容量のデータの転送も速くスムーズに登録が進む.
また,ヒト個人を特定する可能性のある遺伝学的なデータおよび表現型の情報に関しては,DDBJのJGA(Japanese Genotype-phenotype Archive)に登録される.これには,科学技術振興機構バイオサイエンスデータベースセンター(National Bioscience Database Center:NBDC)において認可された利用制限ポリシーをもつ匿名化されたデータのみが登録できる.NBDCではヒトに関するさまざまなデータを共有するためのプラットフォーム“NBDCヒトデータベース”を運用しており(http://humandbs.biosciencedbc.jp/),次世代シークエンサーにより得られたデータに関してはJGAに登録されるしくみになっている.
現時点ではデータを蓄積するほうに重点がおかれているように思うかもしれないが,今後,公共データベースに蓄積されたデータを再利用した研究が増えていくに違いない23).そのため,きたるべき利用に備えて,どういった実験をしたか,そのデータを発表した論文はどれかなど,配列データそのものだけでなく,実験に関するデータ(メタデータ)もきちんと登録し今後の科学の発展に貢献できるよう心がけてほしい.
公共データベースSRAには,すでに“次々世代”シークエンサーから得られたデータも登録されている.しかしながら,現状ではシークエンサーの種類が変わってもデータ解析の手順が大きく変わることは考えられない.すなわち,データ解析の手法としてはある程度が固まってきた感がある.今後は,インターフェースの改良などによりデータ解析におけるハードルを下げる努力がなされ,生物学者自身が次世代シークエンサーにより得られたデータを解析する時代になっていくことだろう.
略歴:2003年 京都大学大学院理学研究科にて博士号取得,理化学研究所ゲノム科学総合研究センター 基礎科学特別研究員,埼玉医科大学ゲノム医学研究センター 助手,同 講師,同 助教授を経て,2007年よりライフサイエンス統合データベースセンター 特任准教授.
© 2015 坊農 秀雅 Licensed under CC 表示 2.1 日本
(ライフサイエンス統合データベースセンター)
email:坊農秀雅
領域融合レビュー, 4, e008 (2015) DOI: 10.7875/leading.author.4.e008
Hidemasa Bono: Sequence data analysis in life science utilizing next generation sequencers.
要 約
生命科学の研究において次世代シークエンサーが普通に使われるようになってきた.これまで,さまざまな応用が提案されてきたが,最近では,ゲノムの再解読による多型の解析やゲノムの新規な解読,トランスクリプトームの解読によるRNA転写量の測定,DNA結合タンパク質の結合配列の解析,細菌叢のメタゲノムの解析がおもなものになった.対応するデータ解析の手法もほぼ固まってきたようにみえる.そこで,このレビューでは,次世代シークエンサーにより得られたデータの解析手法を,公共データベースのデータを解析してきた立場から紹介する.
はじめに
次世代シークエンサー(next generation sequencer:NGS)により解読された塩基配列の情報は,どのような実験を行ったかというメタデータとともに,SRA(Sequence Read Archive)とよばれる公共データベースに登録されている1).次世代シークエンサーにより得られたデータの登録は2007年からはじまり,2015年4月現在,総塩基数で約3.6ペタ塩基(ペタは10の15乗),データ量は約2.3ペタバイトと,保持するだけでもたいへんな量になっている(http://www.ncbi.nlm.nih.gov/Traces/sra/).その研究分野による内訳をみると,ゲノムが3/4近くをしめ,その残りの半分がトランスクリプトーム,ついでメタゲノムなっている(図1).このレビューでは,次世代シークエンサーにより得られたデータの解析手法を解説する.
1.マッピングとアセンブル
次世代シークエンサーにより得られたデータの解析に用いられるソフトウェアの多くはオープンソースで無償で使えるものであり,多くのユーザーがそれをテストし各種のメーリングリストやtwitterなどのソーシャルメディアでその評判が流布している.それらをまとめたSEQanswersのWikiには,2014年4月15日現在,690ものソフトウェアが登録されている(http://seqanswers.com/wiki/Software).数多くのソフトウェアが存在するものの,やっていること自体はほぼ同じというものが多く,ソフトウェアの種類自体はほぼ出尽くした感がある.それらのうち代表的なものを紹介する.これら次世代シークエンサーに関連する配列データのフォーマットをまとめた(表1).
次世代シークエンサーから直接に得るにしても,SRAなどの公共データベースからダウンロードするにしても,データ解析のハブはFASTQ形式の配列ファイルである(図2).そのFASTQファイルをもとに,データを解析する前処理としてアダプター配列やタグ配列を除去し品質管理を行うが,その目的にはFASTQCというソフトウェアがよく用いられる(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).そののち,データ解析はリファレンスとなるゲノムにマッピングするか,アセンブルするかに分かれる.
リファレンス配列がすでにあるヒトや,ゲノム配列がすでに解読されているマウス,ショウジョウバエ,線虫などの多くの古典的なモデル生物においては,次世代シークエンサーにより得られたリード配列をリファレンスとなるゲノム配列に対して“貼りつけ”(マッピング)をすることからデータ解析がはじまる.このマッピングのためのソフトウェアとしては,Bowtie 2) あるいはBWA 3) が使われることがほとんどである.マッピングには大きな計算コストがかかり,多くの計算時間およびメモリを要する.その出力結果はゲノムに対するBAM形式のアラインメントファイルとして得られる.Bowtieの出力結果はBAM形式のテキスト版であるSAM形式により得られるが,そののちのデータ処理の入力がソートされたBMA形式であることが多いので,samtoolsというソフトウェアを用いてBAM形式に変換しソートすることが多い4).最新版のsamtools(version1.2)ではこのソートの並列化が実装され変換の高速化が図られている.
リファレンスとなるゲノム配列のない生物種ではマッピングはできないなので,次世代シークエンサーにより得られたリード配列の“つなぎあわせ”(アセンブル)をする必要がある.アセンブルにより得られるのは,BLASTなど配列類似性の検索でおなじみのFASTA形式の配列データである.
2.ゲノムの解析
次世代シークエンサーがもっとも使われているのは,個体のあいだのゲノムの解析,とくに,ヒト個人のゲノム解読である.ヒトゲノム全体の1%ほどのmRNAに転写されるエキソン領域のみを再解読の対象とするエキソーム解析では,マッピングのためのソフトウェアとしてBWAを使う解析フローが紹介されることが多い.その一例を簡単に述べると,マッピングにより得られたBAMファイルから,samtoolsやPicard(http://picard.sourceforge.net/)を使い重複のあるリード配列を除き,Bedtoolsを用いてエキソン領域のみを抽出する5).そして,samtoolsにより多型(とくに1塩基置換,single nucleotide variant:SNV)のある場所を抽出し,非同義置換,ミスセンス変異,フレームシフト変異というアノテーションをつけ,VCF形式のファイルとして結果を得る.
また,エピゲノムを解析する場合には,バイサルファイト処理によりメチル化されなかったシトシンがウラシルに置換されDNA配列を解読する際にチミンとして読まれることを利用してメチル化された部位を見い出すWGBS(whole genome bisulfite sequencing)法により解析する.メチル化された部位に塩基置換が起こるのでそれを1塩基置換として見い出す戦略をとり,基本的なデータ解析の手法はゲノムの再解読のときと同じである.最終的には,ゲノムブラウザとしてよく使われるIGV 6) を用いて,候補となる領域を研究者が自分の目で確認する.
また,ゲノムを新規に解読する場合にはアセンブルの必要があるが,そのためのソフトウェア(アセンブラー)は多く開発されており,たとえば,nucleotid.esというWebサイト(http://nucleotid.es/)にカタログ化されている.なかでも,米国Broad Instituteにおいて開発されたALLPATH-LG 7) や,日本発のPlatanus 8) などがよく使われている.これらは無償で利用できるが,アセンブルのためのソフトウェアは一般に大きなメモリが必要となるので個人や研究室の所有するマシンでは動かせないことが多い.そこで,スーパーコンピューター(スパコン)を利用することになるが,研究目的なら国立遺伝学研究所のスパコンにおいてさまざまなソフトウェアが試用できるので利用するとよい.また,有償のソフトウェアとしては,デンマークのCLC bio社が開発しているCLC assembly cell(http://www.clcbio.com/products/clc-assembly-cell/)はスパコンにしかない大きなメモリを必要とせずMacOSXでも実行が可能で,さまざまな理由から外部に出せない配列データのアセンブルに適している.アセンブルによりコンティグが得られたら,それらの順序および向きをそろえてより長い配列を得る必要がある.それをやってくれるのがOperaというソフトウェアである9).まず,FASTA形式のコンティグのファイルとそれを生成するのに使ったリード配列のFASTQ形式のファイル,マッピングに使うソフトウェア(BWAあるいはBowtie)を引数にあたえて実行し,map形式の結果ファイルを得る.そののち,Operaを起動してFASTA形式の配列ファイルを得ることにより,より少なく,かつ,平均的により長くなったコンティグが得られる.得らえたゲノム配列は,近縁種のcDNAやアミノ酸配列に対する配列類似性検索によりアノテーションし,最終的にはGTF形式(GFF形式)のファイルを得る.
3.トランスクリプトームの解析
マイクロアレイを用いたハイブリダイゼーション法をベースにした手法が主流であったRNA転写量の測定も,次世代シークエンサーを用いたRNA-seq法がとって代わろうとしている.これは,転写されたRNAの配列をすべて解読し,それぞれの個数をそれが由来する転写単位(遺伝子)の発現強度とする手法である.かつて,Bodymap法とよばれる手法では,EST(expressed sequence tag)とよばれるmRNAの配列断片をクラスタリングし,転写単位ごとにその数を数えあげることにより遺伝子発現量を解析していた10).RNA-seq法は,まさに次世代シークエンサーを使うことによりこのBodymap法をなしとげるものである.RNA-seq法により得られるRNA転写量の単位として,RPKM(reads per kilobase per million mapped reads)がよく用いられる.これは正規化された遺伝子発現量で,100万個のリード配列をマッピングし転写産物の長さを1000塩基としたときのマッピングされたリード配列の数である11).また,RPKMの代わりに使われることの多いFPKM(fragments per kilobase of exon per million mapped)もほぼ同じで,断片ごとの正規化された遺伝子発現量である.RNA-seq法に関しても,リファレンスとなるゲノムにマッピングするかアセンブルするかに分かれる.
マッピングによる方法では,エキソーム解析と同じくBowtieやBWAというマッピングのためのソフトウェアが使われる.しかし,RNAに特有のスプライシングに対するアラインメントが必要になる.それを行うのがTopHatというソフトウェアで,TopHatが内部でBowtieを起動するため,RNA-seq法ではマッピングにBowtieが使われることが多い12).そののち,ゲノムのどの位置に遺伝子があるかなどを記述したゲノムアノテーションのファイル(多くの場合,GTF形式)を使い,Cufflinksというソフトウェアにより選択的スプライシングによるスプライスバリアントをリストアップする13).Cufflinksにより出力される遺伝子発現量はFPKMである.Cufflinksは複数のソフトウェアからなり,いくつかの計算ステップが必要ではあるが,Cuffdiffにより指定した2つの状態の遺伝子発現量の差を同定することができる.さらに,Cufflinksの結果を読み込んでR/Bioconductorにおいて便利に使えるようにするcummeRbundというパッケージもある.トランスクリプトーム解析のための多くのソフトウェアがR/Bioconductorで開発されていることもあり,さらなるデータ解析が進めやすく便利である14).ただし,TopHat(Bowtie)を実行したのちにCufflinksを実行するという一連の過程は必要なメモリの量が多く,多くのCPUが搭載されているマシンであっても1つのCPUで実行される部分もあるなど,計算には数時間のオーダーがかかる.そこで,より高速なソフトウェアの開発が進められ,最近では,アラインメントをせずにk-merをカウントすることにより遺伝子発現を定量する方法が注目されている.その代表的なソフトウェアにSailfishがある15).このソフトウェアはトランスクリプトームが既知でないと利用できないが,いちどインデックスを作成さえしておけば,あとはFASTQファイルごとにかなり高速に遺伝子発現が定量できる.
アセンブルによる方法では,Trinityというソフトウェアがよく使われる16).必要なメモリの量が多く計算時間も長くかかるのが難点であるが,国立遺伝学研究所のスパコンでも試用できる.
ここまでの手順により遺伝子単位で発現量を定量してしまえば,マイクロアレイ解析での手順がほぼそのまま利用できる.抽出した遺伝子セットが遺伝子全体からみてどういう特徴をもつのかみるのによく用いられるのがGSEA(gene set enrichment analysis)法である17).もちろん,エキソーム解析やのちに述べるChIP-seq法などにおいてもこのGSEA法は有効である.なかでも,DAVIDとよばれるウェブツールはインターフェースもよくできており便利である18).OMIM,Gene Ontology,Pathwayという機能情報への遺伝子アノテーションを利用したデータの解釈は,マイクロアレイにおけるデータ解析と同様に,次世代シークエンサーにより得られた大量のデータを解釈する手段として有効である(図3).さらに最近,論文データベースであるPubMedにおいておのおのの論文に付与されているMeSH(medical subject headings)を使いGSEA法により解析する手法を実装したR/Bioconductorのパッケージmeshrが公開され,データ解析のバリエーションがさらに広がった19).
4.DNA結合タンパク質の結合配列の解析
以前より,DNA結合タンパク質の結合したDNA配列を解析する手法としてクロマチン免疫沈降(chromatin immunoprecipitation:ChIP)法があり,2000年代前半から2000年代中ごろにかけては,DNAの配列断片をマイクロアレイにより検出するChIP-on-chip法が用いられていた.そして,マイクロアレイの代わりに次世代シークエンサーを用いてDNAの配列断片を解読する方法が開発されChIP-seq法とよばれるようになった20).ChIP-seq法では,DNA結合タンパク質を認識する抗体を用いてこれが結合したDNAの配列断片を回収し解析する.ターゲットとなるタンパク質としてヒストンと転写因子がある.ともに結合したDNA配列を解読することによりゲノムのどの領域に結合していたかを知ることができ,ゲノムのどの位置に遺伝子があるかというゲノムアノテーション情報とつきあわせることにより直接的な転写制御の関係が推定できる.
ChIP-seq法においては,クロマチン免疫沈降法によりDNA結合タンパク質に結合したDNAの配列断片をリファレンスとなるゲノムにマッピングし,得られたBAM形式のアラインメントファイルを入力として,MACSというソフトウェアを使い結合部位を推定する21).得られる結果は,染色体の番号,その場所(startとend)とその場所でのピークの値がかかれたBED形式のファイルである.この情報からゲノムのどこにピークがあるかがわかる.そのゲノムにおける位置がどこか,ゲノムアノテーション情報をもとにR/Bioconductorなどを使い解析する.ChIP-seq法におけるデータ解析にあたりむずかしいのは,“結合がある”とするかどうかの閾値の線引きで,こればかりは実際のデータをみて個々に決めていく必要がある.また,得られた結合配列の特徴(転写因子の場合には,転写因子結合配列モチーフ)を知りたい場合には,それらの配列を抽出しアラインメントしたのち,その配列の特徴をWebLogoを用いて頻出する塩基が大きく表示されるよう可視化するなどの手段がある22).
5.メタゲノムの解析
次世代シークエンサーにより細菌叢の全体がもつDNAの配列をいちどに解読できるようになり,ショットガン法により細菌叢の全ゲノムを解析する方法や,16S rRNAのみを解読し細菌叢に存在する各種の細菌の割合を解析する方法が開発されている.その結果,ヒト腸内細菌叢のメタゲノムは,ヒト,マウスについで公共データベースSRAに多く登録されている.16S rRNAを解析する方法は,解読されたリード配列に含まれる16S rRNAの配列がデータベース化された既知の16S rRNAの配列のどれにマッチするかを探索するものである.全ゲノムの解析については,微生物における全ゲノム配列の解読と同じステップをふむ.まず,解読されたリード配列を既知のデータベースにたよることなくアセンブルする.その配列からタンパク質の配列をコードするORFを見い出し,得られたアミノ酸配列を質問配列として,これまでに知られているアミノ酸配列のデータベースに対しBLASTなどを使い配列類似性を検索する.その結果から,配列類似性にもとづき機能アノテーションし,最終的に,KEGGやCOGという分類のためのデータベースを用いて機能分類をする.
6.データの再利用
次世代シークエンサーにより得られたデータは基本的には公共データベースであるSRAに登録される.それは,解析結果の再現性の担保のため,そして,科学の進展のためであり,現在,われわれが思いもよらないような使い方がされそこから大発見があるかもしれないからである.例をあげるなら,かつて理化学研究所のFANTOMプロジェクトにおいて作製されたESTデータベースは,iPS細胞の分化を誘導する4つの因子を絞り込むことに使われた(http://www.osc.riken.jp/contents/fantom/).今後も同じようなことが起こるよう,次世代シークエンサーにより得られたデータは公共データベースへ登録していくべきなのである.わが国のDDBJ(DNA Data Bank of Japan)は米国のNCBIや欧州のEBIとデータを交換しているので,DDBJのDRA(DDBJ Sequence Read Archive)に登録してもSRAに登録するのと同じであり,大容量のデータの転送も速くスムーズに登録が進む.
また,ヒト個人を特定する可能性のある遺伝学的なデータおよび表現型の情報に関しては,DDBJのJGA(Japanese Genotype-phenotype Archive)に登録される.これには,科学技術振興機構バイオサイエンスデータベースセンター(National Bioscience Database Center:NBDC)において認可された利用制限ポリシーをもつ匿名化されたデータのみが登録できる.NBDCではヒトに関するさまざまなデータを共有するためのプラットフォーム“NBDCヒトデータベース”を運用しており(http://humandbs.biosciencedbc.jp/),次世代シークエンサーにより得られたデータに関してはJGAに登録されるしくみになっている.
現時点ではデータを蓄積するほうに重点がおかれているように思うかもしれないが,今後,公共データベースに蓄積されたデータを再利用した研究が増えていくに違いない23).そのため,きたるべき利用に備えて,どういった実験をしたか,そのデータを発表した論文はどれかなど,配列データそのものだけでなく,実験に関するデータ(メタデータ)もきちんと登録し今後の科学の発展に貢献できるよう心がけてほしい.
おわりに
公共データベースSRAには,すでに“次々世代”シークエンサーから得られたデータも登録されている.しかしながら,現状ではシークエンサーの種類が変わってもデータ解析の手順が大きく変わることは考えられない.すなわち,データ解析の手法としてはある程度が固まってきた感がある.今後は,インターフェースの改良などによりデータ解析におけるハードルを下げる努力がなされ,生物学者自身が次世代シークエンサーにより得られたデータを解析する時代になっていくことだろう.
文 献
- Kodama, Y., Shumway, M. & Leinonen, R.: The Sequence Read Archive: explosive growth of sequencing data. Nucleic Acids Res., 40, D54-D56 (2012)[PubMed]
- Langmead, B. & Salzberg, S. L.: Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9, 357-359 (2012)[PubMed]
- Li, H. & Durbin, R.: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754-1760 (2009)[PubMed]
- Li, H., Handsaker, B., Wysoker, A. et al.: The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078-2079 (2009)[PubMed]
- Quinlan, A. R. & Hall, I. M.: BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841-842 (2010)[PubMed]
- Robinson, J. T., Thorvaldsdottir, H., Winckler, W. et al.: Integrative genomics viewer. Nat. Biotechnol., 29, 24-26 (2011)[PubMed]
- Gnerre, S., Maccallum, I., Przybylski, D. et al.: High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA, 108, 1513-1518 (2011)[PubMed]
- Kajitani, R., Toshimoto, K., Noguchi, H. et al.: Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res., 24, 1384-1395 (2014)[PubMed]
- Gao, S., Sung, W. K. & Nagarajan, N.: Opera: reconstructing optimal genomic scaffolds with high-throughput pair-end sequences. J. Comput. Biol., 18, 1681-1691 (2011)[PubMed]
- Okubo, K., Hori, N., Matoba, R. et al.: Large scale cDNA sequencing for analysis of quantitative and qualitative aspects of gene expression. Nat. Genet., 3, 173-179 (1992)[PubMed]
- Mortazavi, A., Williams, B. A., McCue, K. et al.: Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods, 7, 621-628 (2008)[PubMed]
- Kim, D., Pertea, G., Trapnell, C. et al.: TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol., 14, R36 (2013)[PubMed]
- Trapnell, C., Hendrickson, D. G., Sauvageau, M. et al.: Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol., 31, 46-53 (2013)[PubMed]
- 門田幸二: トランスクリプトーム解析. 共立出版 (2014)
- Patro, R., Mount, S. M. & Kingford, C.: Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat. Biotechnol., 32, 462-464 (2014)[PubMed]
- Grabherr, M. G., Haas, B. J., Yassour, M. et al.: Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol., 29, 644-652 (2011)[PubMed]
- Subramanian, A., Tamayo, P., Mootha, V. K. et al.: Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA, 102, 15545-15550 (2005)[PubMed]
- Huang, D. W., Sherman, B. T. & Lempicki, R. A.: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc., 4, 44-57 (2009)[PubMed]
- Tsuyuzaki, K., Morota, G., Ishii, M. et al.: MeSH ORA framework: R/Bioconductor packages to support MeSH over-representation analysis. BMC Bioinformatics, 16, 45 (2015)[PubMed]
- Kharchenko, P. V., Tolstorukov, M. Y. & Park, P. J.: Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol., 26, 1351-1359 (2008)[PubMed]
- Zhang, Y., Liu, T., Meyer, C. A. et al.: Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137 (2008)[PubMed]
- Crooks, G. E., Hon, G., Chandonia, J. M. et al.: WebLogo: a sequence logo generator. Genome Res., 14, 1188-1190 (2004)[PubMed]
- Second call for pan-cancer analysis. Nat. Genet., 46, 1251 (2014)[PubMed]
著者プロフィール
略歴:2003年 京都大学大学院理学研究科にて博士号取得,理化学研究所ゲノム科学総合研究センター 基礎科学特別研究員,埼玉医科大学ゲノム医学研究センター 助手,同 講師,同 助教授を経て,2007年よりライフサイエンス統合データベースセンター 特任准教授.
© 2015 坊農 秀雅 Licensed under CC 表示 2.1 日本
