小胞体におけるタンパク質の品質管理の分子機構および中枢神経系における小胞体ストレス応答の役割
2015/06/02
西頭 英起
(宮崎大学医学部 機能生化学分野)
email:西頭英起
領域融合レビュー, 4, e009 (2015) DOI: 10.7875/leading.author.4.e009
Hideki Nishitoh: The molecular mechanisms of endoplasmic reticulum quality control and the roles of unfolded protein response in central nervous system.

細胞が正常に機能するには,遺伝情報にしたがい合成されたタンパク質が正しい高次構造をとることが必要不可欠である.しかし,化学的,物理的,遺伝的なさまざまなストレスにより正しい高次構造をとっていない不良なタンパク質が小胞体に蓄積することは細胞の正常な機能のさまたげになる.このような危機的な状態を回避するため,細胞はUPRとよばれる巧妙なストレス応答による小胞体におけるタンパク質の品質管理機構をもち,それにより不良タンパク質は正しい機能を発揮する.しかし,小胞体に過度の不良タンパク質が蓄積しUPRでは対処しきれなくなったとき,あるいは,細胞がうけるストレスによりUPRが破綻したときにはアポトーシスが誘導される.異常な構造をもつタンパク質の蓄積を原因とするコンホメーション病の分子機構として,小胞体ストレスにより誘導される細胞機能の低下やアポトーシスの関与が注目されている.一方,最近では,さまざまな臓器におけるUPRの生理的な役割も明らかにされつつあり,UPRは生体の機能に必須のストレス応答であることがわかってきた.このレビューでは,小胞体においてタンパク質はどのように品質管理されているのか,また,小胞体ストレス応答の役割について,中枢神経系に焦点をしぼり解説する.
細胞が新たに合成するタンパク質の多くは正しい高次構造をとっていない不良タンパク質であるが,その約1/3は翻訳ののちすぐにユビキチン-プロテアソーム系により分解される1).一方,翻訳の途中のポリソームの約8割は脂質膜により構成されるなんらかのオルガネラと結合しており2),すべてのタンパク質の約1/3は小胞体を通過するともいわれている.したがって,リボソームの多くは小胞体膜あるいはその近傍にて翻訳を行っており,そこで合成されるタンパク質のかなりの割合が不良タンパク質として小胞体に負荷をかけていると考えられる.真核生物の細胞において合成された分泌タンパク質および膜タンパク質は,小胞体において高次構造が形成され機能的なタンパク質となる.しかし,細胞がさまざまなストレスにさらされ不良タンパク質が小胞体に蓄積すると小胞体ストレスという状態におちいる.細胞はこのバランスの乱れた状態を感知し恒常性を維持するためのタンパク質の品質管理機構として大きく2つの手段をとる(図1).ひとつは,小胞体に挿入される新規のタンパク質の合成を抑制することにより小胞体にかかる負荷を軽減する手段である.この機構においておもに寄与するのは,細胞の全体における翻訳の抑制,および,小胞体に挿入されるタンパク質をコードするmRNAを分解するRIDD(regulated IRE1-dependent decay)である.また,タンパク質のトランスロコンを介した小胞体への挿入を特異的に抑制し,細胞質においてこれを分解することにより小胞体へのさらなる負荷を軽減させる予防的な品質管理機構(pre-emptive quality control:pQC)についても報告されている3,4).もうひとつは,すでに合成された不良タンパク質を処理する手段である.すなわち,小胞体シャペロンのATP活性を用いてタンパク質をふたたび折りたたみ正しい高次構造を形成させるか,あるいは,もはやそれが不可能な不良タンパク質については細胞質に逆輸送してこれを分解するERAD(endoplasmic reticulum-associated degradation,小胞体関連分解)である.これらのような小胞体におけるタンパク質の品質管理機構は総じて広義にUPR(unfolded protein response)とよばれる.ところが,UPRによるストレスの回避能をこえるような小胞体ストレスが負荷された場合やタンパク質の品質管理機構が低下した場合にはアポトーシスのシグナルが発信される.小胞体ストレスは小胞体膜に存在する受容体により感知され,そのシグナルは小胞体の外へと伝達される.これまで,受容体が小胞体ストレスを認識し活性化する分子機構や,下流のエフェクターおよびシグナル伝達の分子機構の多くが明らかにされている.このレビューでは,これらUPRの分子機構を説明するとともに,その役割のひとつとして,中枢神経系におけるUPRの生理的な意義とその破綻による疾患との関係について最新の知見をまとめる.
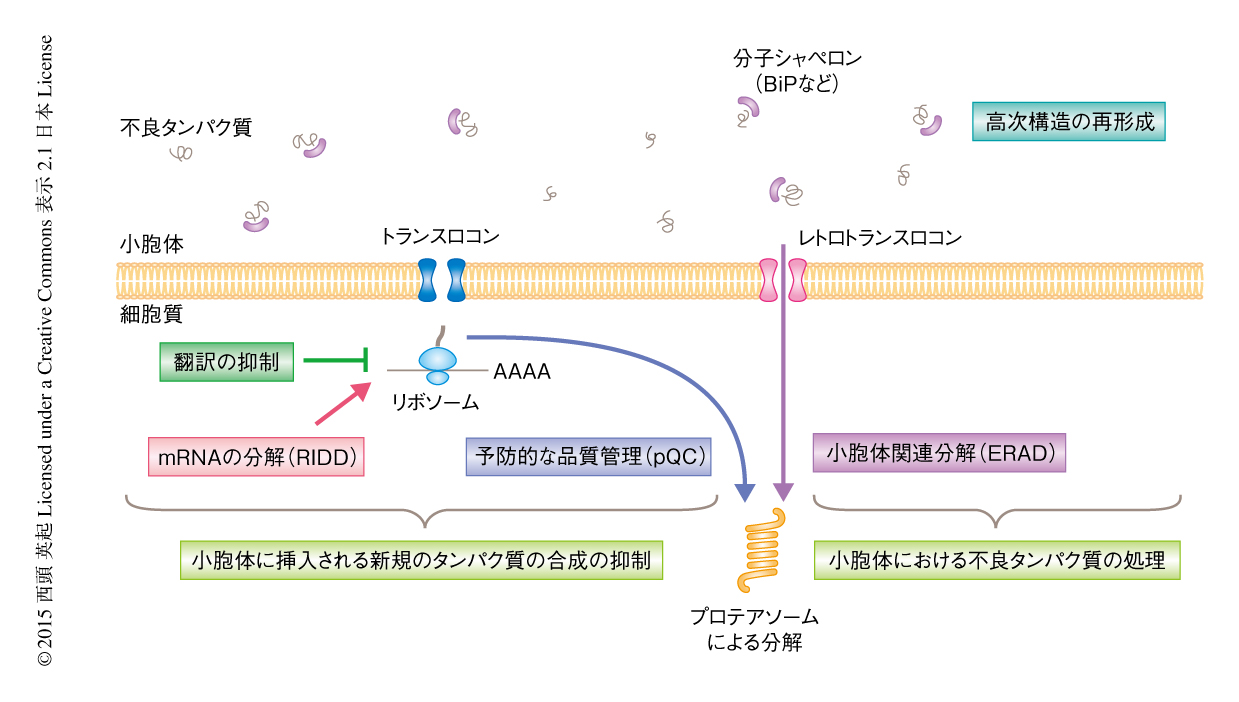
脊椎動物において,小胞体における不良タンパク質の蓄積あるいは小胞体膜を構成する脂質の変化は,ユニークな活性化の機構をもつ3種類の受容体,PERK,IRE1,ATF6により認識される.これらはいずれも膜貫通型のタンパク質であり,定常状態では小胞体内腔側に分子シャペロンBiP/GRP78が結合して不活性型として維持されている.小胞体における不良タンパク質の蓄積によりBiPが受容体から不良タンパク質へと移行することがきっかけとなり,これらの受容体は活性化され下流にシグナルが伝達される.
PERKは細胞質側にセリン/スレオニンキナーゼドメインをもつ.小胞体ストレスを感知したPERKは二量体化さらにオリゴマー化するにともないキナーゼドメインが自己リン酸化し,高次構造を変化させることにより活性型になる5)(図2).活性型のPERKは基質であるeIF2αを特異的にリン酸化する.真核生物における翻訳の多くはキャップ依存的な翻訳,すなわち,Met-tRNA,GTP,eIF2α-eIFβ-eIFγからなる三者複合体が5’側の末端にキャップ構造をもつmRNAと翻訳開始因子eIF4Fとの複合体と結合することにより開始する.eIF2αのリン酸化により三者複合体の形成が阻害されることでキャップ依存的な翻訳は抑制され,細胞の全体における翻訳の速度は低下し,小胞体に挿入される新規のタンパク質の合成が減少する6).これにより,小胞体へのさらなる負荷は軽減される.一方で,リン酸化されたeIF2αはuORF構造をもつ遺伝子の翻訳を逆に促進する.この特殊な分子機構により発現の誘導された転写因子ATF4は,アミノ酸代謝の促進や活性酸素の軽減などにかかわる遺伝子の転写を誘導する7).この機構は統合的ストレス応答(integrated stress response)とよばれ,酸化ストレスや低酸素ストレスなどとも共通する応答である.ところが,このPERK-ATF4経路はアポトーシスの促進にかかわる転写因子,CHOPおよびGADD153の発現を誘導することにより,小胞体ストレスの強度あるいは時間などに応じてアポトーシスのシグナルも誘導する.
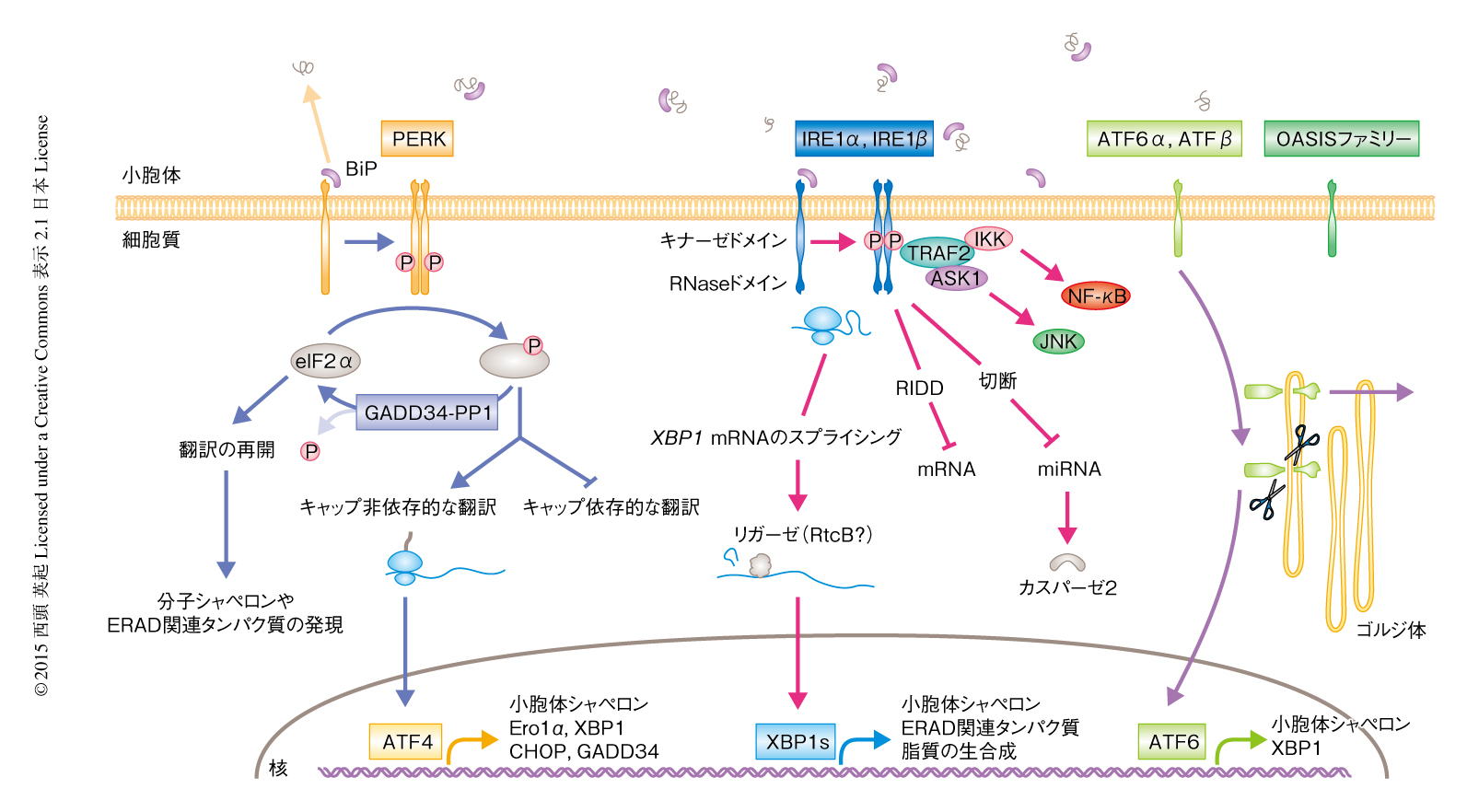
IRE1は細胞質側にキナーゼドメインおよびRNaseドメインの両方をもつ特徴的な構造をもち,哺乳類にはIRE1αおよびIRE1βが存在する.IRE1αはほぼすべての臓器また細胞種に発現しており,IRE1βの発現は腸管系などに限定され,とくに杯細胞におけるムチンの産生に必須である8).PERKと同様に,IRE1も小胞体ストレスを感知するとBiPの解離ののち二量体化し,キナーゼドメインが自己リン酸化し高次構造が変化することによりRNaseドメインが活性化される.哺乳類においては,活性型のRNaseドメインはその特異的な基質であるXBP1 mRNAの2カ所を切断し,26塩基のイントロンがスプライシングされたのち切断されたエキソンが連結される9-11)(図2).XBP1 mRNAの効率的なスプライシングはリボソームと共役して合成されたXBP1タンパク質のポリペプチド鎖が小胞体膜にリクルートされることにより可能になる12,13).エキソンを連結するリガーゼについては長いあいだ不明であったが,最近,RtcBが報告された14).スプライシングされたXBP1 mRNAは転写因子XBP1sをコードしており,ERADに関連するタンパク質や酸化還元酵素などを発現させる.また,オリゴマー化したIRE1は分泌タンパク質や膜タンパク質をコードするmRNAを切断しその発現を抑制する.この機構はさきに述べたようにRIDDとよばれ15,16),小胞体ストレスにより誘導されるアポトーシスとの関連が示唆されている17).持続的に活性化されたIRE1はmRNAのみならず,miR-17,miR-34a,miR-96,miR-125bを選択的に切断する18).これらのマイクロRNAはカスパーゼ2の発現の制御にかかわっており,その結果,カスパーゼ経路の活性化を介してアポトーシスを促進するBcl-2ファミリータンパク質がミトコンドリアへと移行することによりアポトーシスが誘導される.一方,RNase活性に非依存的なIRE1の機能として,NF-κB経路やJNK経路の活性化がある.キナーゼドメインの活性化に依存してアダプタータンパク質TRAF2がIRE1へとリクルートされ19),ASK1を介してJNK経路を活性化しアポトーシスが誘導される20).炎症応答においてはIRE1αとTRAF2との結合を介しIKKのリン酸化およびp65サブユニットの分解が亢進されNF-κB経路が活性化される21).このように,IRE1αは小胞体ストレスの状況に応じてさまざまなシグナル伝達経路を活性化することにより,生存と機能さらにはアポトーシスという多様な細胞の運命決定を担っており,ストレスの強さと長さに応じキナーゼドメインとRNaseドメインの基質がどのように認識されるのか興味深い.
ATF6はC末端側が小胞体内腔側をむいた膜貫通型のタンパク質で,そのN末端にbZIPドメインをもつ膜型の転写因子である.脊椎動物にはATF6αおよびATF6βが存在し,それぞれが代償性に機能する22).ATF6は小胞体ストレスに依存してゴルジ体へと輸送され,そこに存在するsite-1プロテアーゼおよびsite-2プロテアーゼにより膜の内部において切断をうける23)(図2).切断されたN末端側の断片は核へと移行し,BiPなどの分子シャペロンの転写を誘導することにより小胞体におけるタンパク質の高次構造の形成を向上させる.これまで,ATF6と類似の構造をもつ5種類のOASISファミリータンパク質,OASIS/CREB3L1,BBF2H7/CREB3L2,CREBH/CREB3L3,AIbZIP/CREB3L4/Tisp40/CREB4,Luman/LZiP/CREB3が同定されている.いずれも,ゴルジ体において膜の内部において切断をうけN末端側の断片が転写因子として機能する.これらは細胞種に特異的な発現を示すものが多く,OASISは骨芽細胞24),アストロサイト25,26),大腸の杯細胞27),BBF2H7は軟骨芽細胞28),脳29),CREBHは肝臓30),CREB4は精巣31,32) などに発現し,その分化や機能において重要な役割をはたしている.最近,OASISファミリーの新たな機能として,BBF2H7のC末端側の断片が細胞の外へと分泌され発生にかかわるヘッジホッグシグナル伝達経路を活性化させ細胞の増殖に関与することが報告された28).このように,ATF6ファミリーはゴルジ体において膜の内部での切断をうけるというユニークな分子機構により活性化されるが,その多様な生物学的な意義に関するさらなる解析が期待される.
細胞がストレスをうけた際に小胞体においてタンパク質の品質を維持するため最初にとる戦略は,PERKによるeIF2αのリン酸化を介した細胞の全体における翻訳の抑制である.しかし,持続的な翻訳の抑制は細胞の生存にとり不都合なため,リン酸化したeIF2αの下流において発現の誘導される転写因子ATF4によりGADD34が発現し,PP1とともにeIF2αを脱リン酸化する33)(図2).この負のフィードバック機構による一過的な翻訳の抑制がのちの小胞体シャペロンやERADに関連するタンパク質の発現の誘導などを可能にする.一方,翻訳の際に小胞体に挿入されるタンパク質には小胞体シャペロンなどのほかにも多くの分泌タンパク質がある.これらシグナル配列をもつタンパク質は定常状態では小胞体に挿入されるが,小胞体ストレスのもとではタンパク質の品質管理に不要なタンパク質をなるべく合成しないようにするため,小胞体への挿入を特異的に阻害して細胞質において直接に分解する予防的な品質管理機構が存在する3,4)(図1).この予防的な品質管理機構の破綻がプリオン病の病態などと関連することが報告されており,中枢神経系におけるタンパク質の品質管理および細胞の機能の維持に必須であることが示唆されているが34),これにかかわる分子機構については現時点では不明である.
UPRによる3つの経路,PERK-eIF2α-ATF4経路,IRE1-XBP1経路,ATF6経路は,BiPなどの小胞体シャペロンのほかにも,酸化還元酵素,レクチンタンパク質,ERADに関連するタンパク質など,小胞体におけるタンパク質の品質管理にかかわるさまざまなタンパク質の発現を誘導する.小胞体に挿入されたタンパク質は,シグナル配列の切断,糖鎖の付加にひきつづき,ジスルフィド結合の形成などの翻訳後修飾が行われる.小胞体は細胞質に比べ酸化状態であり,このことはジスルフィド結合を介した高次構造の形成に重要である.この小胞体の内部の環境を厳密に維持するため,Ero1やPDIなど約20種類の酸化還元酵素の発現の量や相互作用が制御されている35).また,糖タンパク質の品質管理にはカルネキシンおよびカルレティキュリンがレクチン型の分子シャペロンとして酸化異性化酵素ERp57と協調して高次構造の形成にかかわる36,37).このように,小胞体では,分子シャペロン,酸化還元酵素,レクチンタンパク質など高次構造の形成にかかわる多くのタンパク質が,さまざまな種類の不良タンパク質の折りたたみの異常を認識し,分子シャペロンのATPに依存的なシャペロン活性や酸化異性化活性を利用して正しい立体構造を形成している.しかし,立体構造の形成がどうしてもうまく進まない場合や,遺伝子変異によりアミノ酸変異がある場合などには不良タンパク質はその分解経路であるERAD経路へと送り込まれる.立体構造の形成か分解かの分別を担うタンパク質としては,還元酵素ERdj5 38-40) やレクチンタンパク質EDEM 41) などが重要な役割をはたす(図3).
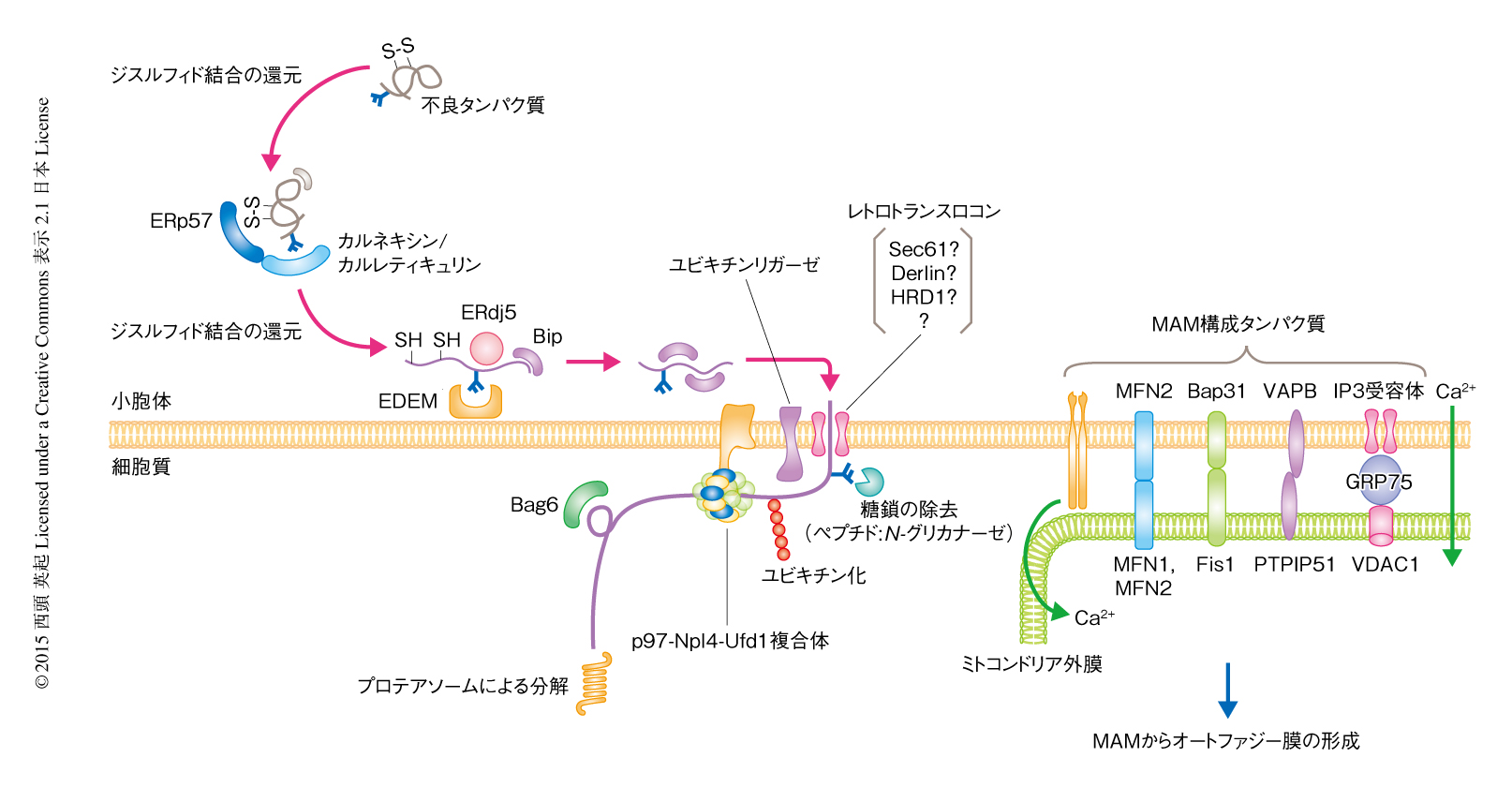
小胞体にタンパク質の分解系は存在しないため,立体構造の形成の不可能な不良タンパク質はERAD経路により小胞体から細胞質へと逆輸送される.この際に不良タンパク質が通過するレトロトランスロコンについては,Sec61トランスロコンであるとする説や42),Derlinファミリータンパク質43,44) やユビキチンリガーゼHRD1 45) などERADに関連するタンパク質が孔を形成しているとする説,あるいは,電子顕微鏡による細胞膜の表面の観察によりトランスロコンのほかには孔らしき構造物が観察されないことから逆輸送孔は存在しないのではないかという仮説もあり46),その実態はいまだ混沌としている.細胞質に放出された不良タンパク質はペプチド:N-グリカナーゼにより糖鎖が除かれたのち47),ユビキチン-プロテアソーム系により分解される48).ERADにより分解されるタンパク質は,高次構造に異常のある領域に応じてERAD-C(細胞質),ERAD-L(小胞体),ERAD-M(小胞体膜)に大別される49).出芽酵母においては,ERAD-CにはユビキチンリガーゼDoa10が,ERAD-LにはユビキチンリガーゼHrd1pが,ERAD-MにはDoa10とHrd1pの両者が関与することが遺伝学的に示されている.しかし,哺乳類においては,HRD1/Synoviolin(出芽酵母Hrd1p),gp78/AMFR/RNF45,RMA1/RNF5,TEB4/MARCH6(出芽酵母Doa10),TMEM129,RNF103/Kf-1,RNF126,RNF138/Trc8,RNF170,RFP2/TRIM13など,およそ10種類のユビキチンリガーゼが関与するが50),それぞれぞれの役割の違いについてはさらに詳細な検討が必要である.ユビキチン化されたタンパク質はDerlinファミリータンパク質に結合するp97(出芽酵母Cdc48)とその補因子であるNpl4とUfd1との複合体を介し,p97のATP加水分解活性に依存してプロテアソームへと輸送される43,51).その際,分解されるタンパク質の疎水性領域が細胞質で凝集しその分解がさまたげられることがないよう,シャペロン様の活性をもつBag6が疎水性領域と結合することにより効率的な分解に寄与している52)(図3).このような一連の機構により不良タンパク質は小胞体や細胞質において凝集することなくスムーズに処理され,細胞は小胞体ストレスを回避して生存し機能することが可能になる.しかし,なんらかの原因によりこれらの分解系が破綻すると細胞の機能の低下や細胞死につながり,さまざまな疾患の原因になる.
小胞体にはタンパク質の合成がさかんな粗面小胞体や脂質の合成の場として機能する滑面小胞体をはじめ,さまざまなサブドメインが存在する.小胞体は核膜からはじまり細胞の全体に広がる構造であり,ほかのオルガネラと接触する分子機構や接触点の形成される生理的な意義が注目されている.とくに,ミトコンドリアと接触している領域はMAM(mitochondria-associated membrane)とよばれ,オートファゴソーム膜の形成53),および,インフラマソームの形成54) における中心的な領域である.MAMは複数のタンパク質により2つのオルガネラ膜が橋渡しされることにより形成される(図3).Ca2+の流入や脂質の供給および代謝など,細胞の機能の維持において重要な役割をはたす.最近,PERKがMAMに局在することが見い出され,MAMの形成およびミトコンドリアにおけるCa2+恒常性の維持に寄与していることが明らかにされたが,その詳細な分子機構は不明である55,56).今後は,UPRのMAMにおける生理的な役割およびその分子機構の解明が期待される.
不良タンパク質の蓄積だけでなく,細胞の膜脂質の恒常性の破綻が小胞体ストレスを誘導しさまざまな疾患に関与することが明らかにされている.膜リン脂質の飽和と不飽和とのバランスがくずれることによりPERKおよびIRE1αは選択的に活性化される57-59).膜脂質の構成の変化によるUPRの活性化はまったく異なる分子機構による.小胞体内腔ドメインを欠失させたPERKおよびIRE1αも膜脂質の飽和化により活性化されたことから58),膜脂質の変化は膜貫通ドメインの近傍において感知されていると予想されるが,詳細な分子機構は不明である.また,膜脂質の飽和化によるUPRシグナルと不良タンパク質の蓄積によるUPRシグナルとは質的に異なるのか,もしそうであれば,その分子機構に関しても興味深い.脂肪の代謝と代謝性疾患との関連を視野に,今後,さらに脂質とUPRの関連についての研究が進むことが期待される.
細胞がUPRにより回避できないような過度の小胞体ストレスをうけた場合,臓器さらには個体の維持のため異常な機能を発揮しないように細胞は自ら死にいたる.このときの細胞死は,小胞体における不良タンパク質の蓄積にひきつづきミトコンドリアの形態に変化が起こり,シトクロムcの放出やカスパーゼ3の活性化などが観察され,最終的にTUNEL陽性を示す核の断片化が誘導される.この細胞死はカスパーゼの阻害剤により抑制されることから,小胞体ストレスにより誘導される細胞死はアポトーシスであるといえる.以下,小胞体ストレスにより誘導されるおもなアポトーシス経路について記述する(図4).
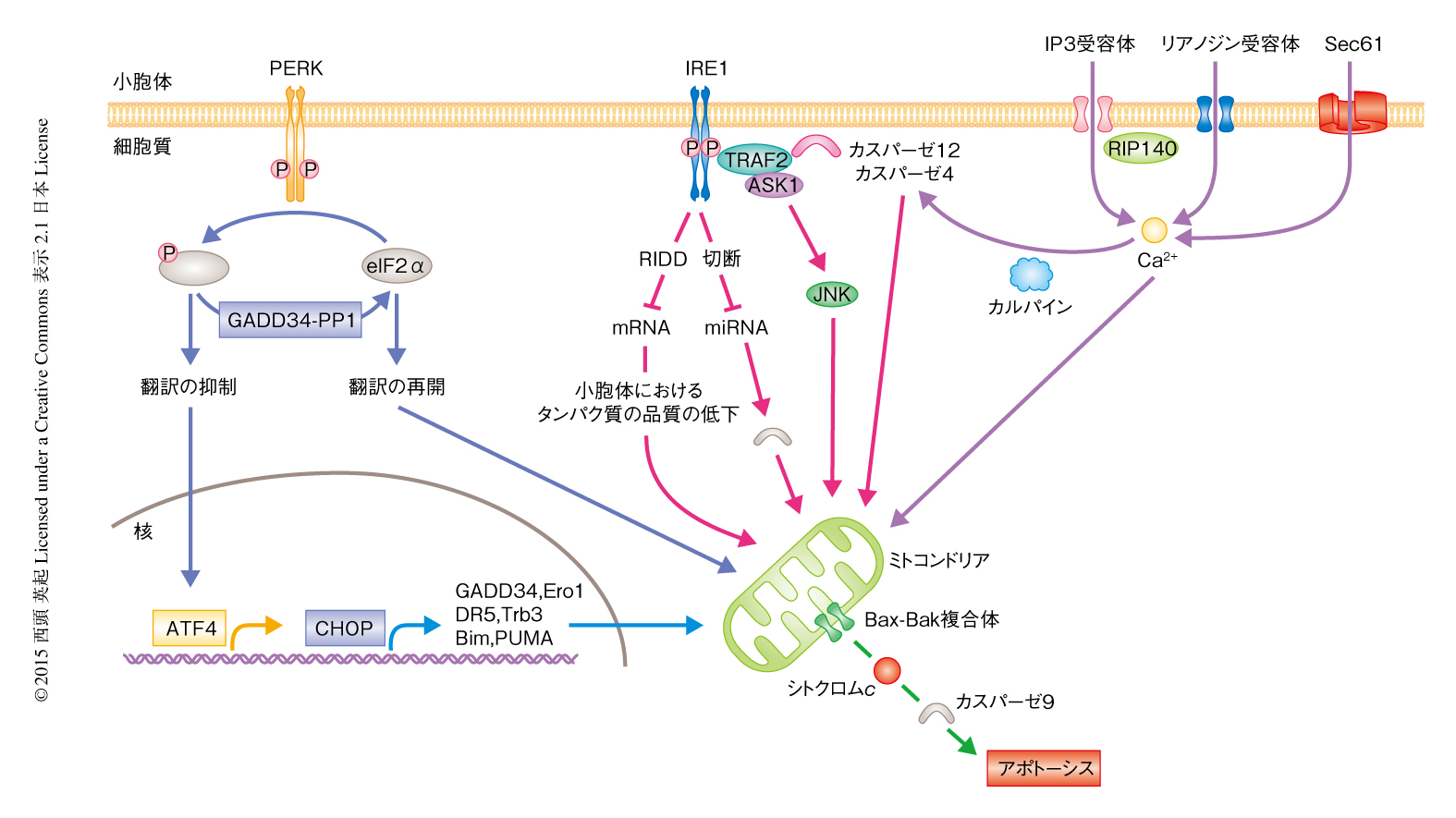
小胞体ストレスにより誘導されるアポトーシスに関連するタンパク質として最初に報告されたのがCHOPである.CHOPはPERK-ATF4経路およびATF6経路の下流において発現の誘導される転写因子のひとつで,標的となる遺伝子の産物としては,GADD34,Ero1,DR5 60),Trb3 61),Bim 62),PUMA 63) などがあり,その多くがアポトーシスに関連した機能をもつ.CHOP欠損細胞やGADD34欠損細胞では小胞体ストレスに依存的なアポトーシスが抑制されることから64-66),この経路の重要性は明らかである.このほかにもCHOPの標的は多く報告されており,ストレスに依存的,また,細胞種に特異的な遺伝子発現を介してアポトーシスが誘導されていると考えられる67).
小胞体ストレスにより誘導されるアポトーシスに関与するカスパーゼは,カスパーゼ2,カスパーゼ3,カスパーゼ4,カスパーゼ7,カスパーゼ8,カスパーゼ9,カスパーゼ12である.最初に注目されたのはマウスのカスパーゼ12で68),ヒトにおけるその機能的なホモログはカスパーゼ4である69).小胞体膜の近傍に局在し,小胞体ストレスに依存的に切断されて活性化型になり細胞質へと移行する.カスパーゼ12欠損細胞では小胞体ストレスに依存的なアポトーシスが顕著に抑制されており,その活性化の機構は,小胞体からイノシトール1,4,5トリスリン酸受容体あるいはリアノジン受容体を介して放出されたCa2+に依存的な活性化カルパインを介してIRE1-TRAF2複合体にカスパーゼ12がリクルートされることによる70,71).活性型カスパーゼ12はBIDの切断およびミトコンドリアからのシトクロムcの放出にともないカスパーゼ9を活性化する.カスパーゼ2およびカスパーゼ8についても同様に,BIDの切断を介してカスパーゼ3およびカスパーゼ7の活性化に依存してアポトーシスを誘導する72,73).カスパーゼ2については,さきに述べたとおり,IRE1に依存的ないくつかのmiRNAの切断により発現が誘導されるというユニークな分子機構が報告されている.一方,カスパーゼ4はカスパーゼ9を直接に切断し活性化する74).このように,多くのカスパーゼファミリーが小胞体ストレスにより誘導されるアポトーシスに関与しているが,これまでは,タプシガルギンやツニカマイシンなど薬剤により誘導された小胞体ストレスにおける研究が多く,今後は,生理的な局面での小胞体ストレスにより誘導されたアポトーシスにおける役割についての研究が重要である.
アポトーシスを促進するBcl-2ファミリータンパク質であるBaxとBakの二重欠損細胞においては,紫外線の刺激などにより誘導されるアポトーシスと同様に,小胞体ストレスにより誘導されるアポトーシスも抑制される.BaxおよびBakは,その作用機序としてミトコンドリア膜のみならず,小胞体膜におけるCa2+恒常性にも直接に関与する75).Bax,イノシトール1,4,5トリスリン酸受容体,Sec61トランスロコンを介して流出したCa2+がセカンドメッセンジャーとしてミトコンドリアに作用しアポトーシスのシグナルが伝達される.転写因子として知られるRIP140は核の外へと移行して小胞体膜のイノシトール1,4,5トリスリン酸受容体と直接に結合することにより小胞体からのCa2+の放出にともなうアポトーシスを負に制御する76).BH-3領域だけをもつBcl-2ファミリータンパク質としては,PUMAおよびNOXAが小胞体ストレスに依存的にp53を介し発現が誘導され,アポトーシスの実行タンパク質として機能する.
IRE1の下流におけるアポトーシスの誘導については,カスパーゼのほかにキナーゼドメインに依存的なASK1-JNK経路がある(図4).一方,持続的に活性化したIRE1はそのRNase活性の特異性が低下し小胞体膜の近傍に存在する多くのmRNAを切断する17).この標的には,分泌タンパク質のほか分子シャペロンなどタンパク質の品質管理に必要なタンパク質のmRNAも含まれ,その結果,小胞体における品質管理機構が低下しアポトーシスが誘導される.
多くの神経変性疾患においては疾患に特異的な異常タンパク質が細胞内あるいは細胞外に蓄積し,神経機能の低下とさらにはニューロンの変性がひき起こされる.このような異常タンパク質の蓄積にも小胞体ストレスが関与する.
アルツハイマー病の患者には脳に異常なタンパク質の蓄積斑がみられ,そのひとつがアミロイドβタンパク質を主要な成分とした細胞外の沈着物である.アルツハイマー病の多くは孤発性であるが一部は家族性であり,その原因遺伝子としてアミロイド前駆体タンパク質,プレセニリン1,プレセニリン2をコードする遺伝子が知られている.いずれの遺伝子変異によってもアミロイドβタンパク質の産生が上昇することなどから,アミロイドβタンパク質の蓄積はアルツハイマー病の発症に関与すると考えられる.アミロイドβタンパク質はアミロイド前駆体タンパク質からβセクレターゼおよびγセクレターゼ(プレセニリ1がその切断活性をもつタンパク質)により切断され細胞の外に分泌される40~42アミノ酸残基からなるペプチドで,神経毒性を発揮しニューロンの変性をひき起こす.培養されたニューロンにおいてアミロイドβタンパク質の添加により細胞死が観察されるが,その際に,カスパーゼ12およびカスパーゼ4が活性化され,これらの欠損細胞においてはアミロイドβタンパク質により誘導されるニューロンの細胞死は抑制される68,69).細胞の外のアミロイドβタンパク質がどのようにして小胞体ストレスを誘導するか,さらに,アルツハイマー病において小胞体ストレスが細胞死をひき起こすことが疾患の発症につながっているのかは議論を残すところである.しかし,病態モデルにおいてXBP1の発現によりアミロイドβタンパク質に依存的に誘導されるニューロンの変性が抑制されることや77),そのほか多くの報告によりアミロイドβタンパク質の神経毒性にUPRが関与することを示唆されていることから,創薬の標的としての可能性もある(図5).
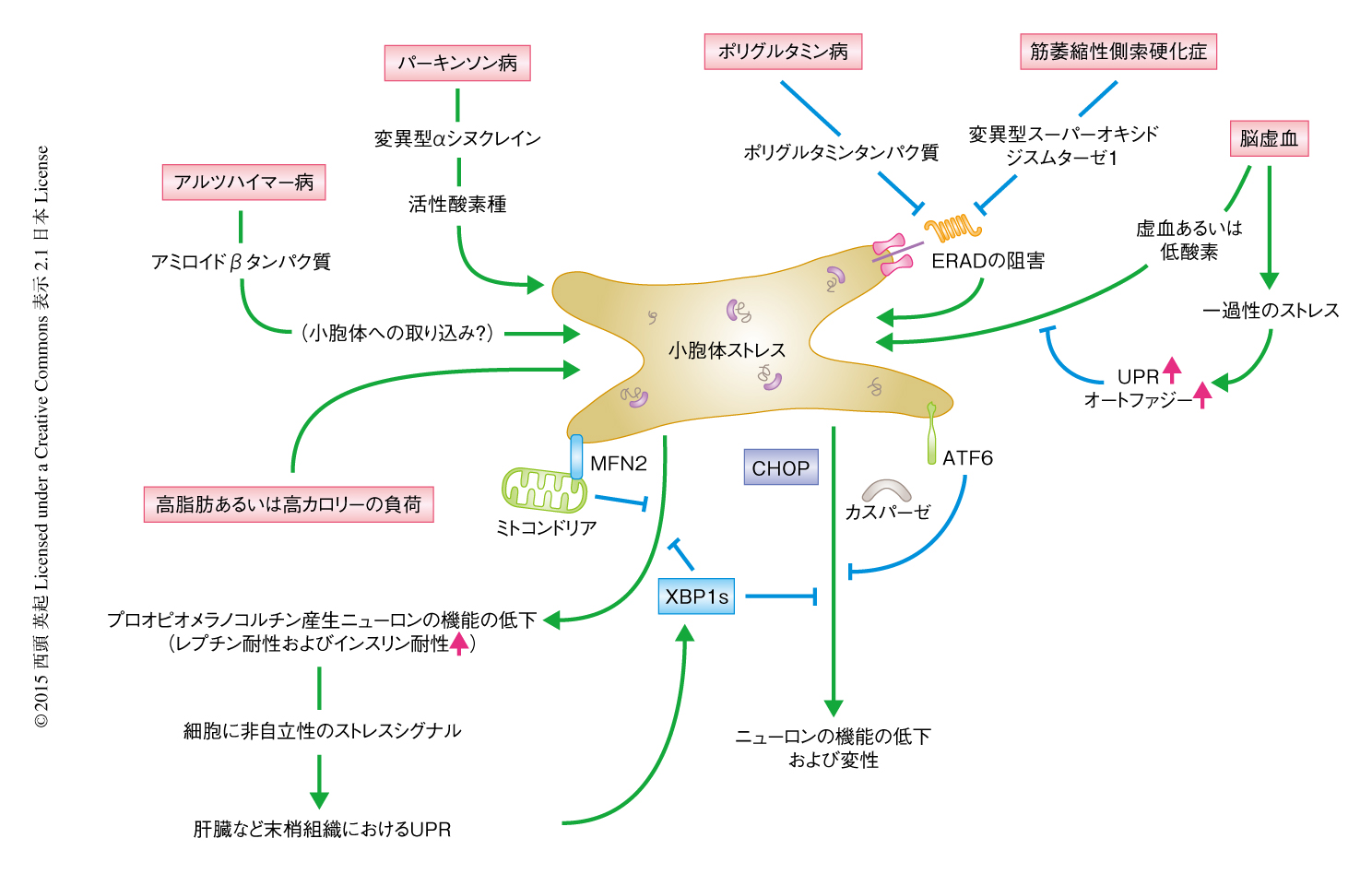
パーキンソン病の患者の中脳黒質や青斑核においてはニューロンの脱落が認められる.孤発性のパーキンソン病の患者において残存したニューロンにはLewy小体とよばれる封入体が観察され,このLewy小体のおもな成分はαシヌクレインである.αシヌクレインは家族性パーキンソン病の原因遺伝子の産物として知られ,PC12細胞においてαシヌクレイン変異体を発現させるとプロテアソームの抑制,シトクロムcの放出,カスパーゼ3およびカスパーゼ9の活性化にくわえ,小胞体ストレスおよびカスパーゼ12の活性化が観察される78).一方で,小胞体ストレスに依存して活性化される転写因子ATF4により,やはりパーキンソン病の原因遺伝子の産物として知られるParkinの発現が誘導され細胞死が抑制される79).1-メチル-4-フェニル-1,2,3,6-テトラヒドロピリジンの脳への投与による慢性パーキンソン病のマウスモデルにおいては,ATF6を介しCHOPの発現が誘導される80).一方,活性化型XBP1sの遺伝子導入はマウスにおいてパーキンソン病の症状を改善する81).このように,パーキンソン病モデルにおいて多くのUPR関連遺伝子の重要性が示されており,創薬の標的として注目されている(図5).しかし,小胞体ストレスがひき起こされる分子機構については活性酸素の関与などが示唆されているが不明な点が多い.また,小胞体ストレスが黒質のドーパミンニューロンのドーパミン分泌能に影響しているのか,また,ニューロンの変性をひき起こしているのか,今後,解明されるべき点は多く残っており,さまざまに存在するパーキンソン病モデルに共通する分子機構の解明が必要である.
ポリグルタミン病は遺伝子においてグルタミンをコードするCAGがくり返し異常に伸長することによりポリグルタミンタンパク質がつくられることに起因する.ポリグルタミンタンパク質はニューロンに封入体を形成し神経機能の障害やニューロンの変性をひき起こす.ハンチントン病,球脊髄性筋萎縮症,Machado-Joseph病などにおいて多くの原因遺伝子が同定されている.ポリグルタミンタンパク質の遺伝子発現への影響やさまざまなタンパク質との結合による神経毒性が報告されているが,そのひとつが,UPSの阻害とそれにともなうユビキチン化タンパク質の蓄積によるニューロンの変性である82-84)(図5).ニューロンにおいてポリグルタミンタンパク質の蓄積によるプロテアソームの抑制は小胞体ストレスを誘導するが20),その分子機構はERADの抑制によると考えられる.UPRとの関連については,XBP1によるポリグルタミンタンパク質の毒性の軽減や85),反対に,PERK-eIF2α経路の必要性が示されている86).ポリグルタミンタンパク質が毒性を発揮するために必要なタンパク質としてショウジョウバエを用いた遺伝学的および生化学的な結合タンパク質の探索により同定されたのがp97である87,88).一方で,ポリグルタミンタンパク質はp97に結合しERADを抑制することにより,小胞体ストレスにより誘導される神経毒性を示すことが報告されている89,90).しかし,p97についてはERADのほかにもさまざまな役割が知られており,その一例として,ポリグルタミンタンパク質との結合が核におけるDNA修復の機能を阻害することがある91).したがって,p97がポリグルタミン病にどのようにかかわるのかについてはいまだ混沌としている.ポリグルタミンタンパク質の分解にはオートファジーも関与することから,細胞に蓄積したポリグルタミンタンパク質がユビキチン-プロテアソーム系だけでなくさまざまな分解経路に影響した結果,小胞体の負荷が増大しニューロンの脆弱性にいたっていることも考えられる.このようなタンパク質の分解能の低下にともなう小胞体ストレスの誘導によるポリグルタミン病の発症機構については未解明な点が残されているが,動物モデルにおいて小胞体ストレスの軽減が疾患の回復につながることが多く報告されていることから,UPRは創薬の分子標的として期待される.
筋萎縮性側索硬化症は運動ニューロンが選択的に脱落することにより起こる重篤な運動機能障害性の疾患である.多くは孤発性であるが約10%が家族性で,その原因となる遺伝子変異の約20%はスーパーオキシドジスムターゼ1をコードするSOD1遺伝子に存在し,これまでに130以上の遺伝子変異が報告されている.変異型SOD1は新たな細胞毒性を獲得することにより運動ニューロンの変性を誘導する.プロテアソーム活性の低下は変異型SOD1の細胞毒性を増強させることから92),晩発性に発症する筋萎縮性側索硬化症の病態機構として加齢などによるタンパク質の分解能の低下と変異型SOD1の蓄積およびその毒性が関与すると予想される.多くの研究グループから,変異型SOD1による神経毒性として小胞体ストレスの関与が報告されている.細胞質に蓄積する変異型SOD1が小胞体に不良タンパク質を蓄積させる分子機構は,ERAD関連タンパク質であるDerlin-1との特異的な結合による.変異型SOD1とDerlin-1との結合はERADにより分解されるタンパク質の逆輸送を阻害し,結果的に,小胞体に不良タンパク質を蓄積させる(図5).このようにしてひき起こされた運動ニューロンおよびその周囲の細胞における小胞体ストレスは,ASK1-JNK経路の活性化,カスパーゼの活性化,CHOPの発現を誘導することにより,運動ニューロンの変性あるいは細胞死をひき起こす93-95).最近,筋萎縮性側索硬化症における運動ニューロンの特異的な脆弱性に関する分子機構について報告された96).障害をうけやすい脊髄前角部の運動ニューロンにおいてはBiPのコシャペロンであるSIL1の発現が低下しており,そのため,小胞体におけるタンパク質の品質管理にかかわるカルレティキュリン,Derlin-1,Rbx1などの発現が低下するため小胞体ストレスに対し脆弱になる.これは,ほぼすべての細胞で発現するSOD1の遺伝子変異がなぜ運動ニューロンを特異的に傷害するか,その分子機構をはじめて示したものである.筋萎縮性側索硬化症と小胞体ストレスとの関係についてはSOD1の遺伝子変異だけでなくTDP-43の遺伝子変異においても報告されており97),家族性のみならず孤発性も含めたすべての筋萎縮性側索硬化症に共通する分子機構として病態に関連する可能性もある.今後は,UPRを標的とした低分子化合物の探索および病態モデルにおける検討が期待される.
以前より,虚血ストレスおよび低酸素ストレスはニューロンにおいて小胞体の機能をいちじるしく障害することが形態学的に観察されてきた98).とくに,海馬CA1領域のニューロンにおいては,一過性の脳虚血ののち小胞体膜におけるタンパク質の合成が阻害され小胞輸送が停止することにより小胞体に不良タンパク質が蓄積し拡大する.このような虚血ストレスおよび低酸素ストレスによる小胞体の機能の低下は中枢神経の障害につながる.CHOPノックアウトマウスでは低酸素による障害ののちのニューロンの変性が軽減され99),反対に,ATF6ノックアウトマウスではグリア細胞の増加をともなうニューロンの変性が増強する100).軽い一過性の脳虚血によりあらかじめUPRを誘導しておきBiPなどの分子シャペロンの発現を増大させることにより,そののちに起こるより大きな侵襲に対しニューロンの耐性を高めることが可能である.また,この軽い一過性の脳虚血によりオートファジーの活性も高まり,2次的な虚血による小胞体ストレスの負荷が軽減される101)(図5).このようなことから,脳の組織においてオートファジーとERADの両方の分解系が機能することにより神経機能の恒常性が保たれていると考えられる.
肥満および糖尿病の病態の機構は,肝臓や膵臓など臓器の機能の異常に起因するだけでなく,中枢神経系から全身へ発信されるペプチドホルモンを介した代謝制御系の破綻も関与することが注目されている.代謝における中枢神経系でのUPRに関しては視床下部においてその重要性が示されている(図5).肥満や糖尿病では,中枢神経性に飽食のシグナルを発する肥満抑制ホルモンであるレプチンやインスリンに対する抵抗性が増大するが,その分子機構は不明であった.視床下部のプロオピオメラノコルチン産生ニューロンにおいては,高脂肪食の負荷によりXBP1sの発現が誘導されることによりレプチン抵抗性およびインスリン抵抗性が緩和される.さらに,プロオピオメラノコルチン産生ニューロンから末梢の肝臓組織へと発信される細胞に非自立性のXBP1sシグナルを介して糖の代謝とインスリン感受性が回復する102).しかし,このような中枢神経系におけるUPRがひとたび破綻すると全身性の代謝異常につながり,肥満あるいは糖尿病の状態が増悪することになる103).また,さきに述べたMAMの構成タンパク質のひとつであるMFN2のプロオピオメラノコルチン産生ニューロンにおける欠失は,小胞体ストレスの増大にともなうレプチン抵抗性による肥満をまねく104).このように,中枢神経とくに視床下部のプロオピオメラノコルチン産生ニューロンにおける小胞体の恒常性の破綻は全身性の代謝の障害につながることから,UPRの細胞に非自立性のシグナル伝達の重要性も注目されている.今後,肥満や糖尿病を含む代謝性疾患における小胞体ストレスの関与についてさらに明らかにされるであろう.
小胞体におけるタンパク質の品質管理機構の解明は,わが国の多くの研究者の貢献によるところが大きい.このレビューでとりあげた以外にも,多様な分子機構が解明されている.しかしその一方で,レトロトランスロコンの実態,IRE1によるXBP1の特異的なスプライシングとRIDDによる幅広いRNAの切断の分子機構の違い,ATF6ファミリーの役割,予防的な品質管理の分子機構など,まだまだ不明な点も多く残されている.また,従来の研究は薬剤により誘導されるUPRの研究が主であったが,近年は,生体における生理的な条件のもとでのストレス応答に関する研究が進められている.このような生理的な小胞体ストレスとその破綻による疾患の分子機構の解明がさらに進展することにより,疾患において具体的な分子標的に対する創薬につながることが期待される.
略歴:1997年 東京医科歯科大学大学院歯学研究科 修了,東京医科歯科大学大学院医歯学総合研究科 助教,同 特任准教授,東京大学大学院薬学系研究科 特任研究員を経て,2012年より宮崎大学医学部 教授.
研究テーマ:オルガネラにおけるストレス応答の機構とその破綻による疾患の分子機構.
研究室URL:http://www.med.miyazaki-u.ac.jp/2bio/index.html
© 2015 西頭 英起 Licensed under CC 表示 2.1 日本
(宮崎大学医学部 機能生化学分野)
email:西頭英起
領域融合レビュー, 4, e009 (2015) DOI: 10.7875/leading.author.4.e009
Hideki Nishitoh: The molecular mechanisms of endoplasmic reticulum quality control and the roles of unfolded protein response in central nervous system.
要 約
細胞が正常に機能するには,遺伝情報にしたがい合成されたタンパク質が正しい高次構造をとることが必要不可欠である.しかし,化学的,物理的,遺伝的なさまざまなストレスにより正しい高次構造をとっていない不良なタンパク質が小胞体に蓄積することは細胞の正常な機能のさまたげになる.このような危機的な状態を回避するため,細胞はUPRとよばれる巧妙なストレス応答による小胞体におけるタンパク質の品質管理機構をもち,それにより不良タンパク質は正しい機能を発揮する.しかし,小胞体に過度の不良タンパク質が蓄積しUPRでは対処しきれなくなったとき,あるいは,細胞がうけるストレスによりUPRが破綻したときにはアポトーシスが誘導される.異常な構造をもつタンパク質の蓄積を原因とするコンホメーション病の分子機構として,小胞体ストレスにより誘導される細胞機能の低下やアポトーシスの関与が注目されている.一方,最近では,さまざまな臓器におけるUPRの生理的な役割も明らかにされつつあり,UPRは生体の機能に必須のストレス応答であることがわかってきた.このレビューでは,小胞体においてタンパク質はどのように品質管理されているのか,また,小胞体ストレス応答の役割について,中枢神経系に焦点をしぼり解説する.
はじめに
細胞が新たに合成するタンパク質の多くは正しい高次構造をとっていない不良タンパク質であるが,その約1/3は翻訳ののちすぐにユビキチン-プロテアソーム系により分解される1).一方,翻訳の途中のポリソームの約8割は脂質膜により構成されるなんらかのオルガネラと結合しており2),すべてのタンパク質の約1/3は小胞体を通過するともいわれている.したがって,リボソームの多くは小胞体膜あるいはその近傍にて翻訳を行っており,そこで合成されるタンパク質のかなりの割合が不良タンパク質として小胞体に負荷をかけていると考えられる.真核生物の細胞において合成された分泌タンパク質および膜タンパク質は,小胞体において高次構造が形成され機能的なタンパク質となる.しかし,細胞がさまざまなストレスにさらされ不良タンパク質が小胞体に蓄積すると小胞体ストレスという状態におちいる.細胞はこのバランスの乱れた状態を感知し恒常性を維持するためのタンパク質の品質管理機構として大きく2つの手段をとる(図1).ひとつは,小胞体に挿入される新規のタンパク質の合成を抑制することにより小胞体にかかる負荷を軽減する手段である.この機構においておもに寄与するのは,細胞の全体における翻訳の抑制,および,小胞体に挿入されるタンパク質をコードするmRNAを分解するRIDD(regulated IRE1-dependent decay)である.また,タンパク質のトランスロコンを介した小胞体への挿入を特異的に抑制し,細胞質においてこれを分解することにより小胞体へのさらなる負荷を軽減させる予防的な品質管理機構(pre-emptive quality control:pQC)についても報告されている3,4).もうひとつは,すでに合成された不良タンパク質を処理する手段である.すなわち,小胞体シャペロンのATP活性を用いてタンパク質をふたたび折りたたみ正しい高次構造を形成させるか,あるいは,もはやそれが不可能な不良タンパク質については細胞質に逆輸送してこれを分解するERAD(endoplasmic reticulum-associated degradation,小胞体関連分解)である.これらのような小胞体におけるタンパク質の品質管理機構は総じて広義にUPR(unfolded protein response)とよばれる.ところが,UPRによるストレスの回避能をこえるような小胞体ストレスが負荷された場合やタンパク質の品質管理機構が低下した場合にはアポトーシスのシグナルが発信される.小胞体ストレスは小胞体膜に存在する受容体により感知され,そのシグナルは小胞体の外へと伝達される.これまで,受容体が小胞体ストレスを認識し活性化する分子機構や,下流のエフェクターおよびシグナル伝達の分子機構の多くが明らかにされている.このレビューでは,これらUPRの分子機構を説明するとともに,その役割のひとつとして,中枢神経系におけるUPRの生理的な意義とその破綻による疾患との関係について最新の知見をまとめる.
1.小胞体ストレスの受容体
脊椎動物において,小胞体における不良タンパク質の蓄積あるいは小胞体膜を構成する脂質の変化は,ユニークな活性化の機構をもつ3種類の受容体,PERK,IRE1,ATF6により認識される.これらはいずれも膜貫通型のタンパク質であり,定常状態では小胞体内腔側に分子シャペロンBiP/GRP78が結合して不活性型として維持されている.小胞体における不良タンパク質の蓄積によりBiPが受容体から不良タンパク質へと移行することがきっかけとなり,これらの受容体は活性化され下流にシグナルが伝達される.
PERKは細胞質側にセリン/スレオニンキナーゼドメインをもつ.小胞体ストレスを感知したPERKは二量体化さらにオリゴマー化するにともないキナーゼドメインが自己リン酸化し,高次構造を変化させることにより活性型になる5)(図2).活性型のPERKは基質であるeIF2αを特異的にリン酸化する.真核生物における翻訳の多くはキャップ依存的な翻訳,すなわち,Met-tRNA,GTP,eIF2α-eIFβ-eIFγからなる三者複合体が5’側の末端にキャップ構造をもつmRNAと翻訳開始因子eIF4Fとの複合体と結合することにより開始する.eIF2αのリン酸化により三者複合体の形成が阻害されることでキャップ依存的な翻訳は抑制され,細胞の全体における翻訳の速度は低下し,小胞体に挿入される新規のタンパク質の合成が減少する6).これにより,小胞体へのさらなる負荷は軽減される.一方で,リン酸化されたeIF2αはuORF構造をもつ遺伝子の翻訳を逆に促進する.この特殊な分子機構により発現の誘導された転写因子ATF4は,アミノ酸代謝の促進や活性酸素の軽減などにかかわる遺伝子の転写を誘導する7).この機構は統合的ストレス応答(integrated stress response)とよばれ,酸化ストレスや低酸素ストレスなどとも共通する応答である.ところが,このPERK-ATF4経路はアポトーシスの促進にかかわる転写因子,CHOPおよびGADD153の発現を誘導することにより,小胞体ストレスの強度あるいは時間などに応じてアポトーシスのシグナルも誘導する.
IRE1は細胞質側にキナーゼドメインおよびRNaseドメインの両方をもつ特徴的な構造をもち,哺乳類にはIRE1αおよびIRE1βが存在する.IRE1αはほぼすべての臓器また細胞種に発現しており,IRE1βの発現は腸管系などに限定され,とくに杯細胞におけるムチンの産生に必須である8).PERKと同様に,IRE1も小胞体ストレスを感知するとBiPの解離ののち二量体化し,キナーゼドメインが自己リン酸化し高次構造が変化することによりRNaseドメインが活性化される.哺乳類においては,活性型のRNaseドメインはその特異的な基質であるXBP1 mRNAの2カ所を切断し,26塩基のイントロンがスプライシングされたのち切断されたエキソンが連結される9-11)(図2).XBP1 mRNAの効率的なスプライシングはリボソームと共役して合成されたXBP1タンパク質のポリペプチド鎖が小胞体膜にリクルートされることにより可能になる12,13).エキソンを連結するリガーゼについては長いあいだ不明であったが,最近,RtcBが報告された14).スプライシングされたXBP1 mRNAは転写因子XBP1sをコードしており,ERADに関連するタンパク質や酸化還元酵素などを発現させる.また,オリゴマー化したIRE1は分泌タンパク質や膜タンパク質をコードするmRNAを切断しその発現を抑制する.この機構はさきに述べたようにRIDDとよばれ15,16),小胞体ストレスにより誘導されるアポトーシスとの関連が示唆されている17).持続的に活性化されたIRE1はmRNAのみならず,miR-17,miR-34a,miR-96,miR-125bを選択的に切断する18).これらのマイクロRNAはカスパーゼ2の発現の制御にかかわっており,その結果,カスパーゼ経路の活性化を介してアポトーシスを促進するBcl-2ファミリータンパク質がミトコンドリアへと移行することによりアポトーシスが誘導される.一方,RNase活性に非依存的なIRE1の機能として,NF-κB経路やJNK経路の活性化がある.キナーゼドメインの活性化に依存してアダプタータンパク質TRAF2がIRE1へとリクルートされ19),ASK1を介してJNK経路を活性化しアポトーシスが誘導される20).炎症応答においてはIRE1αとTRAF2との結合を介しIKKのリン酸化およびp65サブユニットの分解が亢進されNF-κB経路が活性化される21).このように,IRE1αは小胞体ストレスの状況に応じてさまざまなシグナル伝達経路を活性化することにより,生存と機能さらにはアポトーシスという多様な細胞の運命決定を担っており,ストレスの強さと長さに応じキナーゼドメインとRNaseドメインの基質がどのように認識されるのか興味深い.
ATF6はC末端側が小胞体内腔側をむいた膜貫通型のタンパク質で,そのN末端にbZIPドメインをもつ膜型の転写因子である.脊椎動物にはATF6αおよびATF6βが存在し,それぞれが代償性に機能する22).ATF6は小胞体ストレスに依存してゴルジ体へと輸送され,そこに存在するsite-1プロテアーゼおよびsite-2プロテアーゼにより膜の内部において切断をうける23)(図2).切断されたN末端側の断片は核へと移行し,BiPなどの分子シャペロンの転写を誘導することにより小胞体におけるタンパク質の高次構造の形成を向上させる.これまで,ATF6と類似の構造をもつ5種類のOASISファミリータンパク質,OASIS/CREB3L1,BBF2H7/CREB3L2,CREBH/CREB3L3,AIbZIP/CREB3L4/Tisp40/CREB4,Luman/LZiP/CREB3が同定されている.いずれも,ゴルジ体において膜の内部において切断をうけN末端側の断片が転写因子として機能する.これらは細胞種に特異的な発現を示すものが多く,OASISは骨芽細胞24),アストロサイト25,26),大腸の杯細胞27),BBF2H7は軟骨芽細胞28),脳29),CREBHは肝臓30),CREB4は精巣31,32) などに発現し,その分化や機能において重要な役割をはたしている.最近,OASISファミリーの新たな機能として,BBF2H7のC末端側の断片が細胞の外へと分泌され発生にかかわるヘッジホッグシグナル伝達経路を活性化させ細胞の増殖に関与することが報告された28).このように,ATF6ファミリーはゴルジ体において膜の内部での切断をうけるというユニークな分子機構により活性化されるが,その多様な生物学的な意義に関するさらなる解析が期待される.
2.小胞体におけるタンパク質の品質管理機構
細胞がストレスをうけた際に小胞体においてタンパク質の品質を維持するため最初にとる戦略は,PERKによるeIF2αのリン酸化を介した細胞の全体における翻訳の抑制である.しかし,持続的な翻訳の抑制は細胞の生存にとり不都合なため,リン酸化したeIF2αの下流において発現の誘導される転写因子ATF4によりGADD34が発現し,PP1とともにeIF2αを脱リン酸化する33)(図2).この負のフィードバック機構による一過的な翻訳の抑制がのちの小胞体シャペロンやERADに関連するタンパク質の発現の誘導などを可能にする.一方,翻訳の際に小胞体に挿入されるタンパク質には小胞体シャペロンなどのほかにも多くの分泌タンパク質がある.これらシグナル配列をもつタンパク質は定常状態では小胞体に挿入されるが,小胞体ストレスのもとではタンパク質の品質管理に不要なタンパク質をなるべく合成しないようにするため,小胞体への挿入を特異的に阻害して細胞質において直接に分解する予防的な品質管理機構が存在する3,4)(図1).この予防的な品質管理機構の破綻がプリオン病の病態などと関連することが報告されており,中枢神経系におけるタンパク質の品質管理および細胞の機能の維持に必須であることが示唆されているが34),これにかかわる分子機構については現時点では不明である.
UPRによる3つの経路,PERK-eIF2α-ATF4経路,IRE1-XBP1経路,ATF6経路は,BiPなどの小胞体シャペロンのほかにも,酸化還元酵素,レクチンタンパク質,ERADに関連するタンパク質など,小胞体におけるタンパク質の品質管理にかかわるさまざまなタンパク質の発現を誘導する.小胞体に挿入されたタンパク質は,シグナル配列の切断,糖鎖の付加にひきつづき,ジスルフィド結合の形成などの翻訳後修飾が行われる.小胞体は細胞質に比べ酸化状態であり,このことはジスルフィド結合を介した高次構造の形成に重要である.この小胞体の内部の環境を厳密に維持するため,Ero1やPDIなど約20種類の酸化還元酵素の発現の量や相互作用が制御されている35).また,糖タンパク質の品質管理にはカルネキシンおよびカルレティキュリンがレクチン型の分子シャペロンとして酸化異性化酵素ERp57と協調して高次構造の形成にかかわる36,37).このように,小胞体では,分子シャペロン,酸化還元酵素,レクチンタンパク質など高次構造の形成にかかわる多くのタンパク質が,さまざまな種類の不良タンパク質の折りたたみの異常を認識し,分子シャペロンのATPに依存的なシャペロン活性や酸化異性化活性を利用して正しい立体構造を形成している.しかし,立体構造の形成がどうしてもうまく進まない場合や,遺伝子変異によりアミノ酸変異がある場合などには不良タンパク質はその分解経路であるERAD経路へと送り込まれる.立体構造の形成か分解かの分別を担うタンパク質としては,還元酵素ERdj5 38-40) やレクチンタンパク質EDEM 41) などが重要な役割をはたす(図3).
小胞体にタンパク質の分解系は存在しないため,立体構造の形成の不可能な不良タンパク質はERAD経路により小胞体から細胞質へと逆輸送される.この際に不良タンパク質が通過するレトロトランスロコンについては,Sec61トランスロコンであるとする説や42),Derlinファミリータンパク質43,44) やユビキチンリガーゼHRD1 45) などERADに関連するタンパク質が孔を形成しているとする説,あるいは,電子顕微鏡による細胞膜の表面の観察によりトランスロコンのほかには孔らしき構造物が観察されないことから逆輸送孔は存在しないのではないかという仮説もあり46),その実態はいまだ混沌としている.細胞質に放出された不良タンパク質はペプチド:N-グリカナーゼにより糖鎖が除かれたのち47),ユビキチン-プロテアソーム系により分解される48).ERADにより分解されるタンパク質は,高次構造に異常のある領域に応じてERAD-C(細胞質),ERAD-L(小胞体),ERAD-M(小胞体膜)に大別される49).出芽酵母においては,ERAD-CにはユビキチンリガーゼDoa10が,ERAD-LにはユビキチンリガーゼHrd1pが,ERAD-MにはDoa10とHrd1pの両者が関与することが遺伝学的に示されている.しかし,哺乳類においては,HRD1/Synoviolin(出芽酵母Hrd1p),gp78/AMFR/RNF45,RMA1/RNF5,TEB4/MARCH6(出芽酵母Doa10),TMEM129,RNF103/Kf-1,RNF126,RNF138/Trc8,RNF170,RFP2/TRIM13など,およそ10種類のユビキチンリガーゼが関与するが50),それぞれぞれの役割の違いについてはさらに詳細な検討が必要である.ユビキチン化されたタンパク質はDerlinファミリータンパク質に結合するp97(出芽酵母Cdc48)とその補因子であるNpl4とUfd1との複合体を介し,p97のATP加水分解活性に依存してプロテアソームへと輸送される43,51).その際,分解されるタンパク質の疎水性領域が細胞質で凝集しその分解がさまたげられることがないよう,シャペロン様の活性をもつBag6が疎水性領域と結合することにより効率的な分解に寄与している52)(図3).このような一連の機構により不良タンパク質は小胞体や細胞質において凝集することなくスムーズに処理され,細胞は小胞体ストレスを回避して生存し機能することが可能になる.しかし,なんらかの原因によりこれらの分解系が破綻すると細胞の機能の低下や細胞死につながり,さまざまな疾患の原因になる.
3.UPRとミトコンドリア
小胞体にはタンパク質の合成がさかんな粗面小胞体や脂質の合成の場として機能する滑面小胞体をはじめ,さまざまなサブドメインが存在する.小胞体は核膜からはじまり細胞の全体に広がる構造であり,ほかのオルガネラと接触する分子機構や接触点の形成される生理的な意義が注目されている.とくに,ミトコンドリアと接触している領域はMAM(mitochondria-associated membrane)とよばれ,オートファゴソーム膜の形成53),および,インフラマソームの形成54) における中心的な領域である.MAMは複数のタンパク質により2つのオルガネラ膜が橋渡しされることにより形成される(図3).Ca2+の流入や脂質の供給および代謝など,細胞の機能の維持において重要な役割をはたす.最近,PERKがMAMに局在することが見い出され,MAMの形成およびミトコンドリアにおけるCa2+恒常性の維持に寄与していることが明らかにされたが,その詳細な分子機構は不明である55,56).今後は,UPRのMAMにおける生理的な役割およびその分子機構の解明が期待される.
4.膜脂質の変化とUPR
不良タンパク質の蓄積だけでなく,細胞の膜脂質の恒常性の破綻が小胞体ストレスを誘導しさまざまな疾患に関与することが明らかにされている.膜リン脂質の飽和と不飽和とのバランスがくずれることによりPERKおよびIRE1αは選択的に活性化される57-59).膜脂質の構成の変化によるUPRの活性化はまったく異なる分子機構による.小胞体内腔ドメインを欠失させたPERKおよびIRE1αも膜脂質の飽和化により活性化されたことから58),膜脂質の変化は膜貫通ドメインの近傍において感知されていると予想されるが,詳細な分子機構は不明である.また,膜脂質の飽和化によるUPRシグナルと不良タンパク質の蓄積によるUPRシグナルとは質的に異なるのか,もしそうであれば,その分子機構に関しても興味深い.脂肪の代謝と代謝性疾患との関連を視野に,今後,さらに脂質とUPRの関連についての研究が進むことが期待される.
5.小胞体ストレスにより誘導されるアポトーシス
細胞がUPRにより回避できないような過度の小胞体ストレスをうけた場合,臓器さらには個体の維持のため異常な機能を発揮しないように細胞は自ら死にいたる.このときの細胞死は,小胞体における不良タンパク質の蓄積にひきつづきミトコンドリアの形態に変化が起こり,シトクロムcの放出やカスパーゼ3の活性化などが観察され,最終的にTUNEL陽性を示す核の断片化が誘導される.この細胞死はカスパーゼの阻害剤により抑制されることから,小胞体ストレスにより誘導される細胞死はアポトーシスであるといえる.以下,小胞体ストレスにより誘導されるおもなアポトーシス経路について記述する(図4).
小胞体ストレスにより誘導されるアポトーシスに関連するタンパク質として最初に報告されたのがCHOPである.CHOPはPERK-ATF4経路およびATF6経路の下流において発現の誘導される転写因子のひとつで,標的となる遺伝子の産物としては,GADD34,Ero1,DR5 60),Trb3 61),Bim 62),PUMA 63) などがあり,その多くがアポトーシスに関連した機能をもつ.CHOP欠損細胞やGADD34欠損細胞では小胞体ストレスに依存的なアポトーシスが抑制されることから64-66),この経路の重要性は明らかである.このほかにもCHOPの標的は多く報告されており,ストレスに依存的,また,細胞種に特異的な遺伝子発現を介してアポトーシスが誘導されていると考えられる67).
小胞体ストレスにより誘導されるアポトーシスに関与するカスパーゼは,カスパーゼ2,カスパーゼ3,カスパーゼ4,カスパーゼ7,カスパーゼ8,カスパーゼ9,カスパーゼ12である.最初に注目されたのはマウスのカスパーゼ12で68),ヒトにおけるその機能的なホモログはカスパーゼ4である69).小胞体膜の近傍に局在し,小胞体ストレスに依存的に切断されて活性化型になり細胞質へと移行する.カスパーゼ12欠損細胞では小胞体ストレスに依存的なアポトーシスが顕著に抑制されており,その活性化の機構は,小胞体からイノシトール1,4,5トリスリン酸受容体あるいはリアノジン受容体を介して放出されたCa2+に依存的な活性化カルパインを介してIRE1-TRAF2複合体にカスパーゼ12がリクルートされることによる70,71).活性型カスパーゼ12はBIDの切断およびミトコンドリアからのシトクロムcの放出にともないカスパーゼ9を活性化する.カスパーゼ2およびカスパーゼ8についても同様に,BIDの切断を介してカスパーゼ3およびカスパーゼ7の活性化に依存してアポトーシスを誘導する72,73).カスパーゼ2については,さきに述べたとおり,IRE1に依存的ないくつかのmiRNAの切断により発現が誘導されるというユニークな分子機構が報告されている.一方,カスパーゼ4はカスパーゼ9を直接に切断し活性化する74).このように,多くのカスパーゼファミリーが小胞体ストレスにより誘導されるアポトーシスに関与しているが,これまでは,タプシガルギンやツニカマイシンなど薬剤により誘導された小胞体ストレスにおける研究が多く,今後は,生理的な局面での小胞体ストレスにより誘導されたアポトーシスにおける役割についての研究が重要である.
アポトーシスを促進するBcl-2ファミリータンパク質であるBaxとBakの二重欠損細胞においては,紫外線の刺激などにより誘導されるアポトーシスと同様に,小胞体ストレスにより誘導されるアポトーシスも抑制される.BaxおよびBakは,その作用機序としてミトコンドリア膜のみならず,小胞体膜におけるCa2+恒常性にも直接に関与する75).Bax,イノシトール1,4,5トリスリン酸受容体,Sec61トランスロコンを介して流出したCa2+がセカンドメッセンジャーとしてミトコンドリアに作用しアポトーシスのシグナルが伝達される.転写因子として知られるRIP140は核の外へと移行して小胞体膜のイノシトール1,4,5トリスリン酸受容体と直接に結合することにより小胞体からのCa2+の放出にともなうアポトーシスを負に制御する76).BH-3領域だけをもつBcl-2ファミリータンパク質としては,PUMAおよびNOXAが小胞体ストレスに依存的にp53を介し発現が誘導され,アポトーシスの実行タンパク質として機能する.
IRE1の下流におけるアポトーシスの誘導については,カスパーゼのほかにキナーゼドメインに依存的なASK1-JNK経路がある(図4).一方,持続的に活性化したIRE1はそのRNase活性の特異性が低下し小胞体膜の近傍に存在する多くのmRNAを切断する17).この標的には,分泌タンパク質のほか分子シャペロンなどタンパク質の品質管理に必要なタンパク質のmRNAも含まれ,その結果,小胞体における品質管理機構が低下しアポトーシスが誘導される.
6.神経変性疾患における小胞体ストレスの関与
多くの神経変性疾患においては疾患に特異的な異常タンパク質が細胞内あるいは細胞外に蓄積し,神経機能の低下とさらにはニューロンの変性がひき起こされる.このような異常タンパク質の蓄積にも小胞体ストレスが関与する.
アルツハイマー病の患者には脳に異常なタンパク質の蓄積斑がみられ,そのひとつがアミロイドβタンパク質を主要な成分とした細胞外の沈着物である.アルツハイマー病の多くは孤発性であるが一部は家族性であり,その原因遺伝子としてアミロイド前駆体タンパク質,プレセニリン1,プレセニリン2をコードする遺伝子が知られている.いずれの遺伝子変異によってもアミロイドβタンパク質の産生が上昇することなどから,アミロイドβタンパク質の蓄積はアルツハイマー病の発症に関与すると考えられる.アミロイドβタンパク質はアミロイド前駆体タンパク質からβセクレターゼおよびγセクレターゼ(プレセニリ1がその切断活性をもつタンパク質)により切断され細胞の外に分泌される40~42アミノ酸残基からなるペプチドで,神経毒性を発揮しニューロンの変性をひき起こす.培養されたニューロンにおいてアミロイドβタンパク質の添加により細胞死が観察されるが,その際に,カスパーゼ12およびカスパーゼ4が活性化され,これらの欠損細胞においてはアミロイドβタンパク質により誘導されるニューロンの細胞死は抑制される68,69).細胞の外のアミロイドβタンパク質がどのようにして小胞体ストレスを誘導するか,さらに,アルツハイマー病において小胞体ストレスが細胞死をひき起こすことが疾患の発症につながっているのかは議論を残すところである.しかし,病態モデルにおいてXBP1の発現によりアミロイドβタンパク質に依存的に誘導されるニューロンの変性が抑制されることや77),そのほか多くの報告によりアミロイドβタンパク質の神経毒性にUPRが関与することを示唆されていることから,創薬の標的としての可能性もある(図5).
パーキンソン病の患者の中脳黒質や青斑核においてはニューロンの脱落が認められる.孤発性のパーキンソン病の患者において残存したニューロンにはLewy小体とよばれる封入体が観察され,このLewy小体のおもな成分はαシヌクレインである.αシヌクレインは家族性パーキンソン病の原因遺伝子の産物として知られ,PC12細胞においてαシヌクレイン変異体を発現させるとプロテアソームの抑制,シトクロムcの放出,カスパーゼ3およびカスパーゼ9の活性化にくわえ,小胞体ストレスおよびカスパーゼ12の活性化が観察される78).一方で,小胞体ストレスに依存して活性化される転写因子ATF4により,やはりパーキンソン病の原因遺伝子の産物として知られるParkinの発現が誘導され細胞死が抑制される79).1-メチル-4-フェニル-1,2,3,6-テトラヒドロピリジンの脳への投与による慢性パーキンソン病のマウスモデルにおいては,ATF6を介しCHOPの発現が誘導される80).一方,活性化型XBP1sの遺伝子導入はマウスにおいてパーキンソン病の症状を改善する81).このように,パーキンソン病モデルにおいて多くのUPR関連遺伝子の重要性が示されており,創薬の標的として注目されている(図5).しかし,小胞体ストレスがひき起こされる分子機構については活性酸素の関与などが示唆されているが不明な点が多い.また,小胞体ストレスが黒質のドーパミンニューロンのドーパミン分泌能に影響しているのか,また,ニューロンの変性をひき起こしているのか,今後,解明されるべき点は多く残っており,さまざまに存在するパーキンソン病モデルに共通する分子機構の解明が必要である.
ポリグルタミン病は遺伝子においてグルタミンをコードするCAGがくり返し異常に伸長することによりポリグルタミンタンパク質がつくられることに起因する.ポリグルタミンタンパク質はニューロンに封入体を形成し神経機能の障害やニューロンの変性をひき起こす.ハンチントン病,球脊髄性筋萎縮症,Machado-Joseph病などにおいて多くの原因遺伝子が同定されている.ポリグルタミンタンパク質の遺伝子発現への影響やさまざまなタンパク質との結合による神経毒性が報告されているが,そのひとつが,UPSの阻害とそれにともなうユビキチン化タンパク質の蓄積によるニューロンの変性である82-84)(図5).ニューロンにおいてポリグルタミンタンパク質の蓄積によるプロテアソームの抑制は小胞体ストレスを誘導するが20),その分子機構はERADの抑制によると考えられる.UPRとの関連については,XBP1によるポリグルタミンタンパク質の毒性の軽減や85),反対に,PERK-eIF2α経路の必要性が示されている86).ポリグルタミンタンパク質が毒性を発揮するために必要なタンパク質としてショウジョウバエを用いた遺伝学的および生化学的な結合タンパク質の探索により同定されたのがp97である87,88).一方で,ポリグルタミンタンパク質はp97に結合しERADを抑制することにより,小胞体ストレスにより誘導される神経毒性を示すことが報告されている89,90).しかし,p97についてはERADのほかにもさまざまな役割が知られており,その一例として,ポリグルタミンタンパク質との結合が核におけるDNA修復の機能を阻害することがある91).したがって,p97がポリグルタミン病にどのようにかかわるのかについてはいまだ混沌としている.ポリグルタミンタンパク質の分解にはオートファジーも関与することから,細胞に蓄積したポリグルタミンタンパク質がユビキチン-プロテアソーム系だけでなくさまざまな分解経路に影響した結果,小胞体の負荷が増大しニューロンの脆弱性にいたっていることも考えられる.このようなタンパク質の分解能の低下にともなう小胞体ストレスの誘導によるポリグルタミン病の発症機構については未解明な点が残されているが,動物モデルにおいて小胞体ストレスの軽減が疾患の回復につながることが多く報告されていることから,UPRは創薬の分子標的として期待される.
筋萎縮性側索硬化症は運動ニューロンが選択的に脱落することにより起こる重篤な運動機能障害性の疾患である.多くは孤発性であるが約10%が家族性で,その原因となる遺伝子変異の約20%はスーパーオキシドジスムターゼ1をコードするSOD1遺伝子に存在し,これまでに130以上の遺伝子変異が報告されている.変異型SOD1は新たな細胞毒性を獲得することにより運動ニューロンの変性を誘導する.プロテアソーム活性の低下は変異型SOD1の細胞毒性を増強させることから92),晩発性に発症する筋萎縮性側索硬化症の病態機構として加齢などによるタンパク質の分解能の低下と変異型SOD1の蓄積およびその毒性が関与すると予想される.多くの研究グループから,変異型SOD1による神経毒性として小胞体ストレスの関与が報告されている.細胞質に蓄積する変異型SOD1が小胞体に不良タンパク質を蓄積させる分子機構は,ERAD関連タンパク質であるDerlin-1との特異的な結合による.変異型SOD1とDerlin-1との結合はERADにより分解されるタンパク質の逆輸送を阻害し,結果的に,小胞体に不良タンパク質を蓄積させる(図5).このようにしてひき起こされた運動ニューロンおよびその周囲の細胞における小胞体ストレスは,ASK1-JNK経路の活性化,カスパーゼの活性化,CHOPの発現を誘導することにより,運動ニューロンの変性あるいは細胞死をひき起こす93-95).最近,筋萎縮性側索硬化症における運動ニューロンの特異的な脆弱性に関する分子機構について報告された96).障害をうけやすい脊髄前角部の運動ニューロンにおいてはBiPのコシャペロンであるSIL1の発現が低下しており,そのため,小胞体におけるタンパク質の品質管理にかかわるカルレティキュリン,Derlin-1,Rbx1などの発現が低下するため小胞体ストレスに対し脆弱になる.これは,ほぼすべての細胞で発現するSOD1の遺伝子変異がなぜ運動ニューロンを特異的に傷害するか,その分子機構をはじめて示したものである.筋萎縮性側索硬化症と小胞体ストレスとの関係についてはSOD1の遺伝子変異だけでなくTDP-43の遺伝子変異においても報告されており97),家族性のみならず孤発性も含めたすべての筋萎縮性側索硬化症に共通する分子機構として病態に関連する可能性もある.今後は,UPRを標的とした低分子化合物の探索および病態モデルにおける検討が期待される.
以前より,虚血ストレスおよび低酸素ストレスはニューロンにおいて小胞体の機能をいちじるしく障害することが形態学的に観察されてきた98).とくに,海馬CA1領域のニューロンにおいては,一過性の脳虚血ののち小胞体膜におけるタンパク質の合成が阻害され小胞輸送が停止することにより小胞体に不良タンパク質が蓄積し拡大する.このような虚血ストレスおよび低酸素ストレスによる小胞体の機能の低下は中枢神経の障害につながる.CHOPノックアウトマウスでは低酸素による障害ののちのニューロンの変性が軽減され99),反対に,ATF6ノックアウトマウスではグリア細胞の増加をともなうニューロンの変性が増強する100).軽い一過性の脳虚血によりあらかじめUPRを誘導しておきBiPなどの分子シャペロンの発現を増大させることにより,そののちに起こるより大きな侵襲に対しニューロンの耐性を高めることが可能である.また,この軽い一過性の脳虚血によりオートファジーの活性も高まり,2次的な虚血による小胞体ストレスの負荷が軽減される101)(図5).このようなことから,脳の組織においてオートファジーとERADの両方の分解系が機能することにより神経機能の恒常性が保たれていると考えられる.
7.肥満ストレスによるUPRの中枢神経系における役割
肥満および糖尿病の病態の機構は,肝臓や膵臓など臓器の機能の異常に起因するだけでなく,中枢神経系から全身へ発信されるペプチドホルモンを介した代謝制御系の破綻も関与することが注目されている.代謝における中枢神経系でのUPRに関しては視床下部においてその重要性が示されている(図5).肥満や糖尿病では,中枢神経性に飽食のシグナルを発する肥満抑制ホルモンであるレプチンやインスリンに対する抵抗性が増大するが,その分子機構は不明であった.視床下部のプロオピオメラノコルチン産生ニューロンにおいては,高脂肪食の負荷によりXBP1sの発現が誘導されることによりレプチン抵抗性およびインスリン抵抗性が緩和される.さらに,プロオピオメラノコルチン産生ニューロンから末梢の肝臓組織へと発信される細胞に非自立性のXBP1sシグナルを介して糖の代謝とインスリン感受性が回復する102).しかし,このような中枢神経系におけるUPRがひとたび破綻すると全身性の代謝異常につながり,肥満あるいは糖尿病の状態が増悪することになる103).また,さきに述べたMAMの構成タンパク質のひとつであるMFN2のプロオピオメラノコルチン産生ニューロンにおける欠失は,小胞体ストレスの増大にともなうレプチン抵抗性による肥満をまねく104).このように,中枢神経とくに視床下部のプロオピオメラノコルチン産生ニューロンにおける小胞体の恒常性の破綻は全身性の代謝の障害につながることから,UPRの細胞に非自立性のシグナル伝達の重要性も注目されている.今後,肥満や糖尿病を含む代謝性疾患における小胞体ストレスの関与についてさらに明らかにされるであろう.
おわりに
小胞体におけるタンパク質の品質管理機構の解明は,わが国の多くの研究者の貢献によるところが大きい.このレビューでとりあげた以外にも,多様な分子機構が解明されている.しかしその一方で,レトロトランスロコンの実態,IRE1によるXBP1の特異的なスプライシングとRIDDによる幅広いRNAの切断の分子機構の違い,ATF6ファミリーの役割,予防的な品質管理の分子機構など,まだまだ不明な点も多く残されている.また,従来の研究は薬剤により誘導されるUPRの研究が主であったが,近年は,生体における生理的な条件のもとでのストレス応答に関する研究が進められている.このような生理的な小胞体ストレスとその破綻による疾患の分子機構の解明がさらに進展することにより,疾患において具体的な分子標的に対する創薬につながることが期待される.
文 献
- Schubert, U., Anton, L. C., Gibbs, J. et al.: Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature, 404, 770-774 (2000)[PubMed]
- Palade, G.: Intracellular aspects of the process of protein synthesis. Science, 189, 347-358 (1975)[PubMed]
- Oyadomari, S., Yun, C., Fisher, E. A. et al.: Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell, 126, 727-739 (2006)[PubMed]
- Kang, S. W., Rane, N. S., Kim, S. J. et al.: Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell, 127, 999-1013 (2006)[PubMed]
- Bertolotti, A., Zhang, Y., Hendershot, L. M. et al.: Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol., 2, 326-332 (2000)[PubMed]
- Harding, H. P., Zhang, Y. & Ron, D.: Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature, 397, 271-274 (1999)[PubMed]
- Harding, H. P., Zhang, Y., Bertolotti, A. et al.: Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell, 5, 897-904 (2000)[PubMed]
- Tsuru, A., Fujimoto, N., Takahashi, S. et al.: Negative feedback by IRE1β optimizes mucin production in goblet cells. Proc. Natl. Acad. Sci. USA, 110, 2864-2869 (2013)[PubMed]
- Calfon, M., Zeng, H., Urano, F. et al.: IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature, 415, 92-96 (2002)[PubMed]
- Shen, X., Ellis, R. E., Lee, K. et al.: Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell, 107, 893-903 (2001)[PubMed]
- Yoshida, H., Matsui, T., Yamamoto, A. et al.: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell, 107, 881-891 (2001)[PubMed]
- Yanagitani, K., Imagawa, Y., Iwawaki, T. et al.: Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol. Cell, 34, 191-200 (2009)[PubMed]
- Yanagitani, K., Kimata, Y., Kadokura, H. et al.: Translational pausing ensures membrane targeting and cytoplasmic splicing of XBP1u mRNA. Science, 331, 586-589 (2011)[PubMed]
- Lu, Y., Liang, F. X. & Wang, X.: A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell, 55, 758-770 (2014)[PubMed]
- Hollien, J., Lin, J. H., Li, H. et al.: Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol., 186, 323-331 (2009)[PubMed]
- Hollien, J. & Weissman, J. S.: Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science, 313, 104-107 (2006)[PubMed]
- Han, D., Lerner, A. G., Vande Walle, L. et al.: IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell, 138, 562-575 (2009)[PubMed]
- Upton, J. P., Wang, L., Han, D. et al.: IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science, 338, 818-822 (2012)[PubMed]
- Urano, F., Wang, X., Bertolotti, A. et al.: Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science, 287, 664-666 (2000)[PubMed]
- Nishitoh, H., Matsuzawa, A., Tobiume, K. et al.: ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev., 16, 1345-1355 (2002)[PubMed]
- Hu, P., Han, Z., Couvillon, A. D. et al.: Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol., 26, 3071-3084 (2006)[PubMed]
- Ishikawa, T., Okada, T., Ishikawa-Fujiwara, T. et al.: ATF6α/β-mediated adjustment of ER chaperone levels is essential for development of the notochord in medaka fish. Mol. Biol. Cell, 24, 1387-1395 (2013)[PubMed]
- Ye, J., Rawson, R. B., Komuro, R. et al.: ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell, 6, 1355-1364 (2000)[PubMed]
- Murakami, T., Saito, A., Hino, S. et al.: Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat. Cell Biol., 11, 1205-1211 (2009)[PubMed]
- Kondo, S., Murakami, T., Tatsumi, K. et al.: OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat. Cell Biol., 7, 186-194 (2005)[PubMed]
- Saito, A., Kanemoto, S., Kawasaki, N. et al.: Unfolded protein response, activated by OASIS family transcription factors, promotes astrocyte differentiation. Nat. Commun., 3, 967 (2012)[PubMed]
- Asada, R., Saito, A., Kawasaki, N. et al.: The endoplasmic reticulum stress transducer OASIS is involved in the terminal differentiation of goblet cells in the large intestine. J. Biol. Chem., 287, 8144-8153 (2012)[PubMed]
- Saito, A., Kanemoto, S., Zhang, Y. et al.: Chondrocyte proliferation regulated by secreted luminal domain of ER stress transducer BBF2H7/CREB3L2. Mol. Cell, 53, 127-139 (2014)[PubMed]
- Kondo, S., Saito, A., Hino, S. et al.: BBF2H7, a novel transmembrane bZIP transcription factor, is a new type of endoplasmic reticulum stress transducer. Mol. Cell Biol, 27, 1716-1729 (2007)[PubMed]
- Zhang, K., Shen, X., Wu, J. et al.: Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell, 124, 587-599 (2006)[PubMed]
- Nagamori, I., Yabuta, N., Fujii, T. et al.: Tisp40, a spermatid specific bZip transcription factor, functions by binding to the unfolded protein response element via the Rip pathway. Genes Cells, 10, 575-594 (2005)[PubMed]
- Stirling, J. & O'Hare, P.: CREB4, a transmembrane bZip transcription factor and potential new substrate for regulation and cleavage by S1P. Mol. Biol. Cell, 17, 413-426 (2006)[PubMed]
- Ma, Y. & Hendershot, L. M.: Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem., 278, 34864-34873 (2003)[PubMed]
- Rane, N. S., Kang, S. W., Chakrabarti, O. et al.: Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev. Cell, 15, 359-370 (2008)[PubMed]
- Araki, K., Iemura, S., Kamiya, Y. et al.: Ero1-α and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J. Cell Biol., 202, 861-874 (2013)[PubMed]
- Ou, W. J., Cameron, P. H., Thomas, D. Y. et al.: Association of folding intermediates of glycoproteins with calnexin during protein maturation. Nature, 364, 771-776 (1993)[PubMed]
- Ellgaard, L., Molinari, M. & Helenius, A.: Setting the standards: quality control in the secretory pathway. Science, 286, 1882-1888 (1999)[PubMed]
- Ushioda, R., Hoseki, J. & Nagata, K.: Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol. Biol. Cell, 24, 3155-3163 (2013)[PubMed]
- Ushioda, R., Hoseki, J., Araki, K. et al.: ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science, 321, 569-572 (2008)[PubMed]
- Hagiwara, M., Maegawa, K., Suzuki, M. et al.: Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol. Cell, 41, 432-444 (2011)[PubMed] [新着論文レビュー]
- Hosokawa, N., Wada, I., Hasegawa, K. et al.: A novel ER α-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep., 2, 415-422 (2001)[PubMed]
- Wiertz, E. J., Tortorella, D., Bogyo, M. et al.: Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature, 384, 432-438 (1996)[PubMed]
- Ye, Y., Shibata, Y., Yun, C. et al.: A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature, 429, 841-847 (2004)[PubMed]
- Lilley, B. N. & Ploegh, H. L.: A membrane protein required for dislocation of misfolded proteins from the ER. Nature, 429, 834-840 (2004)[PubMed]
- Carvalho, P., Stanley, A. M. & Rapoport, T. A.: Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell, 143, 579-591 (2010)[PubMed]
- Ploegh, H. L.: A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature, 448, 435-438 (2007)[PubMed]
- Huang, C., Harada, Y., Hosomi, A. et al.: Endo-β-N-acetylglucosaminidase forms N-GlcNAc protein aggregates during ER-associated degradation in Ngly1-defective cells. Proc. Natl. Acad. Sci. USA, 112, 1398-1403 (2015)[PubMed]
- Meusser, B., Hirsch, C., Jarosch, E. et al.: ERAD: the long road to destruction. Nat. Cell Biol., 7, 766-772 (2005)[PubMed]
- Carvalho, P., Goder, V. & Rapoport, T. A.: Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell, 126, 361-373 (2006)[PubMed]
- Christianson, J. C. & Ye, Y.: Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol., 21, 325-335 (2014)[PubMed]
- Ye, Y., Shibata, Y., Kikkert, M. et al.: Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA, 102, 14132-14138 (2005)[PubMed]
- Wang, Q., Liu, Y., Soetandyo, N. et al.: A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol. Cell, 42, 758-770 (2011)[PubMed]
- Hamasaki, M., Furuta, N., Matsuda, A. et al.: Autophagosomes form at ER-mitochondria contact sites. Nature, 495, 389-393 (2013)[PubMed] [新着論文レビュー]
- Zhou, R., Yazdi, A. S., Menu, P. et al.: A role for mitochondria in NLRP3 inflammasome activation. Nature, 469, 221-225 (2011)[PubMed]
- Verfaillie, T., Rubio, N., Garg, A. D. et al.: PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ., 19, 1880-1891 (2012)[PubMed]
- Garg, A. D., Krysko, D. V., Verfaillie, T. et al.: A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J., 31, 1062-1079 (2012)[PubMed]
- Volmer, R., van der Ploeg, K. & Ron, D.: Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA, 110, 4628-4633 (2013)[PubMed]
- Kitai, Y., Ariyama, H., Kono, N. et al.: Membrane lipid saturation activates IRE1α without inducing clustering. Genes Cells, 18, 798-809 (2013)[PubMed]
- Ariyama, H., Kono, N., Matsuda, S. et al.: Decrease in membrane phospholipid unsaturation induces unfolded protein response. J. Biol. Chem., 285, 22027-22035 (2010)[PubMed]
- Yamaguchi, H. & Wang, H. G.: CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem., 279, 45495-45502 (2004)[PubMed]
- Ohoka, N., Yoshii, S., Hattori, T. et al.: TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J., 24, 1243-1255 (2005)[PubMed]
- Puthalakath, H., O'Reilly, L. A., Gunn, P. et al.: ER stress triggers apoptosis by activating BH3-only protein Bim. Cell, 129, 1337-1349 (2007)[PubMed]
- Cazanave, S. C., Elmi, N. A., Akazawa, Y. et al.: CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am. J. Physiol. Gastrointest. Liver Physiol., 299, G236-G243 (2010)[PubMed]
- Malhotra, J. D., Miao, H., Zhang, K. et al.: Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA, 105, 18525-18530 (2008)[PubMed]
- Marciniak, S. J., Yun, C. Y., Oyadomari, S. et al.: CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev., 18, 3066-3077 (2004)[PubMed]
- Song, B., Scheuner, D., Ron, D. et al.: Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Invest., 118, 3378-3389 (2008)[PubMed]
- Urra, H., Dufey, E., Lisbona, F. et al.: When ER stress reaches a dead end. Biochim. Biophys. Acta, 1833, 3507-3517 (2013)[PubMed]
- Nakagawa, T., Zhu, H., Morishima, N. et al.: Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature, 403, 98-103 (2000)[PubMed]
- Hitomi, J., Katayama, T., Eguchi, Y. et al.: Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J Cell Biol, 165, 347-356 (2004)[PubMed]
- Nakagawa, T. & Yuan, J.: Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol., 150, 887-894 (2000)[PubMed]
- Yoneda, T., Imaizumi, K., Oono, K. et al.: Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem., 276, 13935-13940 (2001)[PubMed]
- Upton, J. P., Austgen, K., Nishino, M. et al.: Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol. Cell. Biol., 28, 3943-3951 (2008)[PubMed]
- Jimbo, A., Fujita, E., Kouroku, Y. et al.: ER stress induces caspase-8 activation, stimulating cytochrome c release and caspase-9 activation. Exp. Cell Res., 283, 156-166 (2003)[PubMed]
- Yamamuro, A., Kishino, T., Ohshima, Y. et al.: Caspase-4 directly activates caspase-9 in endoplasmic reticulum stress-induced apoptosis in SH-SY5Y cells. J. Pharmacol. Sci., 115, 239-243 (2011)[PubMed]
- Sano, R. & Reed, J. C.: ER stress-induced cell death mechanisms. Biochim. Biophys. Acta, 1833, 3460-3470 (2013)[PubMed]
- Feng, X., Krogh, K. A., Wu, C. Y. et al.: Receptor-interacting protein 140 attenuates endoplasmic reticulum stress in neurons and protects against cell death. Nat. Commun., 5, 4487 (2014)[PubMed]
- Casas-Tinto, S., Zhang, Y., Sanchez-Garcia, J. et al.: The ER stress factor XBP1s prevents amyloid-β neurotoxicity. Hum. Mol. Genet., 20, 2144-2160 (2011)[PubMed]
- Smith, W. W., Jiang, H., Pei, Z. et al.: Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet., 14, 3801-3811 (2005)[PubMed]
- Bouman, L., Schlierf, A., Lutz, A. K. et al.: Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ., 18, 769-782 (2011)[PubMed]
- Hashida, K., Kitao, Y., Sudo, H. et al.: ATF6alpha promotes astroglial activation and neuronal survival in a chronic mouse model of Parkinson's disease. PLoS One, 7, e47950 (2012)[PubMed]
- Valdes, P., Mercado, G., Vidal, R. L. et al.: Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA, 111, 6804-6809 (2014)[PubMed]
- Bence, N. F., Sampat, R. M. & Kopito, R. R.: Impairment of the ubiquitin-proteasome system by protein aggregation. Science, 292, 1552-1555 (2001)[PubMed]
- Orr, H. T.: Beyond the Qs in the polyglutamine diseases. Genes Dev., 15, 925-932 (2001)[PubMed]
- Sherman, M. Y. & Goldberg, A. L.: Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron, 29, 15-32 (2001)[PubMed]
- Zuleta, A., Vidal, R. L., Armentano, D. et al.: AAV-mediated delivery of the transcription factor XBP1s into the striatum reduces mutant Huntingtin aggregation in a mouse model of Huntington's disease. Biochem. Biophys. Res. Commun., 420, 558-563 (2012)[PubMed]
- Leitman, J., Barak, B., Benyair, R. et al.: ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS One, 9, e90803 (2014)[PubMed]
- Higashiyama, H., Hirose, F., Yamaguchi, M. et al.: Identification of ter94, Drosophila VCP, as a modulator of polyglutamine-induced neurodegeneration. Cell Death Differ., 9, 264-273 (2002)[PubMed]
- Hirabayashi, M., Inoue, K., Tanaka, K. et al.: VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ., 8, 977-984 (2001)[PubMed]
- Leitman, J., Hartl, F. U. & Lederkremer, G. Z.: Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun., 4, 2753 (2013)[PubMed]
- Duennwald, M. L. & Lindquist, S.: Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev., 22, 3308-3319 (2008)[PubMed]
- Fujita, K., Nakamura, Y., Oka, T. et al.: A functional deficiency of TERA/VCP/p97 contributes to impaired DNA repair in multiple polyglutamine diseases. Nat. Commun., 4, 1816 (2013)[PubMed]
- Kitamura, A., Inada, N., Kubota, H. et al.: Dysregulation of the proteasome increases the toxicity of ALS-linked mutant SOD1. Genes Cells, 19, 209-224 (2014)[PubMed]
- Nishitoh, H., Kadowaki, H., Nagai, A. et al.: ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev., 22, 1451-1464 (2008)[PubMed]
- Soo, K. Y., Atkin, J. D., Farg, M. et al.: Bim links ER stress and apoptosis in cells expressing mutant SOD1 associated with amyotrophic lateral sclerosis. PLoS One, 7, e35413 (2012)[PubMed]
- Wootz, H., Hansson, I., Korhonen, L. et al.: XIAP decreases caspase-12 cleavage and calpain activity in spinal cord of ALS transgenic mice. Exp. Cell Res., 312, 1890-1898 (2006)[PubMed]
- Filezac de L'Etang, A., Maharjan, N., Cordeiro Brana, M. et al.: Marinesco-Sjogren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat. Neurosci., 18, 227-238 (2015)[PubMed]
- Walker, A. K., Soo, K. Y., Sundaramoorthy, V. et al.: ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS One, 8, e81170 (2013)[PubMed]
- Bando, Y., Katayama, T., Kasai, K. et al.: GRP94 (94 kDa glucose-regulated protein) suppresses ischemic neuronal cell death against ischemia/reperfusion injury. Eur. J. Neurosci., 18, 829-840 (2003)[PubMed]
- Tajiri, S., Oyadomari, S., Yano, S. et al.: Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ., 11, 403-415 (2004)[PubMed]
- Yoshikawa, A., Kamide, T., Hashida, K. et al.: Deletion of Atf6α impairs astroglial activation and enhances neuronal death following brain ischemia in mice. J. Neurochem., 132, 342-353 (2015)[PubMed]
- Sheng, R., Liu, X. Q., Zhang, L. S. et al.: Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy, 8, 310-325 (2012)[PubMed]
- Williams, K. W., Liu, T., Kong, X. et al.: Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab., 20, 471-482 (2014)[PubMed]
- Ozcan, L., Ergin, A. S., Lu, A. et al.: Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab., 9, 35-51 (2009)[PubMed]
- Schneeberger, M., Dietrich, M. O., Sebastian, D. et al.: Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell, 155, 172-187 (2013)[PubMed]
著者プロフィール
略歴:1997年 東京医科歯科大学大学院歯学研究科 修了,東京医科歯科大学大学院医歯学総合研究科 助教,同 特任准教授,東京大学大学院薬学系研究科 特任研究員を経て,2012年より宮崎大学医学部 教授.
研究テーマ:オルガネラにおけるストレス応答の機構とその破綻による疾患の分子機構.
研究室URL:http://www.med.miyazaki-u.ac.jp/2bio/index.html
© 2015 西頭 英起 Licensed under CC 表示 2.1 日本
