腫瘍内不均一性とがんの進化
2016/02/19
新井田厚司1・三森功士2・宮野 悟1
(1東京大学医科学研究所 ヘルスインテリジェンスセンター健康医療計算科学分野,2九州大学病院別府病院 外科)
email:新井田厚司
領域融合レビュー, 5, e003 (2016) DOI: 10.7875/leading.author.5.e003
Atsushi Niida, Koshi Mimori & Satoru Miyano: Intratumor heterogeneity and cancer evolution.

長年,がんは直線的なクローン進化をへて形成される悪性度の高いクローナルな細胞の集団としてとらえられてきた.しかしながら,近年,次世代シークエンサーを用いたゲノム解析により,ひとつの腫瘍のなかにも多数のサブクローンの存在することが明らかにされている.さらに,がんの進化のシミュレーションなどにより,この腫瘍内不均一性は中立進化により生じる可能性が示されている.腫瘍内不均一性はがんの治療抵抗性の根本的な原因である可能性もあり,腫瘍内不均一性を生じるがんの進化のシステムレベルでの原理的な理解は臨床的にも重要である.そのためには,今後,がんの研究において異分野との融合を推進していく必要がある.それをみすえて,このレビューにおいては,この分野のこれまでの知見および期待される展開について解説する.
ひとつの腫瘍のなかにはゲノムの異なる複数のクローンの存在することが知られており,この現象は腫瘍内不均一性とよばれている.腫瘍内不均一性はさまざまなタイプのがんにおいて観察されており,がんの治療抵抗性の一因になっていると考えられる.つまり,腫瘍内不均一性があると,治療感受性のクローンが大多数をしめる腫瘍が治療により縮小しても,治療抵抗性の少数のクローンが残存する可能性があり,やがて,そのようなクローンが増殖して再発にいたることが考えられる.腫瘍内不均一性はがんの進化の過程でクローンが分岐することにより生じると考えられる.
1976年,体細胞変異の獲得および変異を獲得したクローンへの自然選択が段階的に起こることにより,がんは単一の正常な細胞から生じることが提唱された1).つまり,がん細胞の進化は非生殖分裂をする単細胞生物のダーウィン的な進化と同様にとらえることができる.このがんのクローン進化説が提唱されたのち,分子生物学によりがん遺伝子およびがん抑制遺伝子が発見され,1990年,これらの発見とクローン進化説は多段階発がん仮説として統合された2).すなわち,大腸がんの発がんの過程において,APC遺伝子,KRAS遺伝子,TP53遺伝子などに発がんの原因となる複数の変異が順に蓄積することにより,正常な上皮細胞が良性腫瘍をへて悪性腫瘍へと直線的に変化するというモデルが提唱された.それ以降,個別の遺伝子レベルでの実験において腫瘍内不均一性の存在が示唆されてきたものの,直線的なクローンの進化をへて悪性度の高いクローナルな細胞の集団として腫瘍が形成されるというモデルが一般的な発がんの過程のイメージとして受け入れられてきた.
近年,次世代シークエンサー(next generation sequencer:NGS)の登場により,がんにおける体細胞変異の包括的なプロファイル,すなわち,がんゲノムを安価かつ迅速に解読することが可能になり,これまで分子生物学が中心であったがんの研究分野において急速なパラダイムシフトが起こっている.The Cancer Genome Atlas(TCGA)やInternational Cancer Genome Consortium(ICGC)におけるような多数の患者から得た試料において塩基配列を解読するゲノム研究により,その変異ががんの発生や進展の直接的な原因になるドライバー遺伝子が数多く同定され,さまざまながんにおいて変異のカタログが作成されている.また,これらの研究により,同一のがんの患者のあいだでもがんゲノムにいちじるしい不均一性のあることが明らかにされ,個別化医療の必要性がとなえられている3,4).
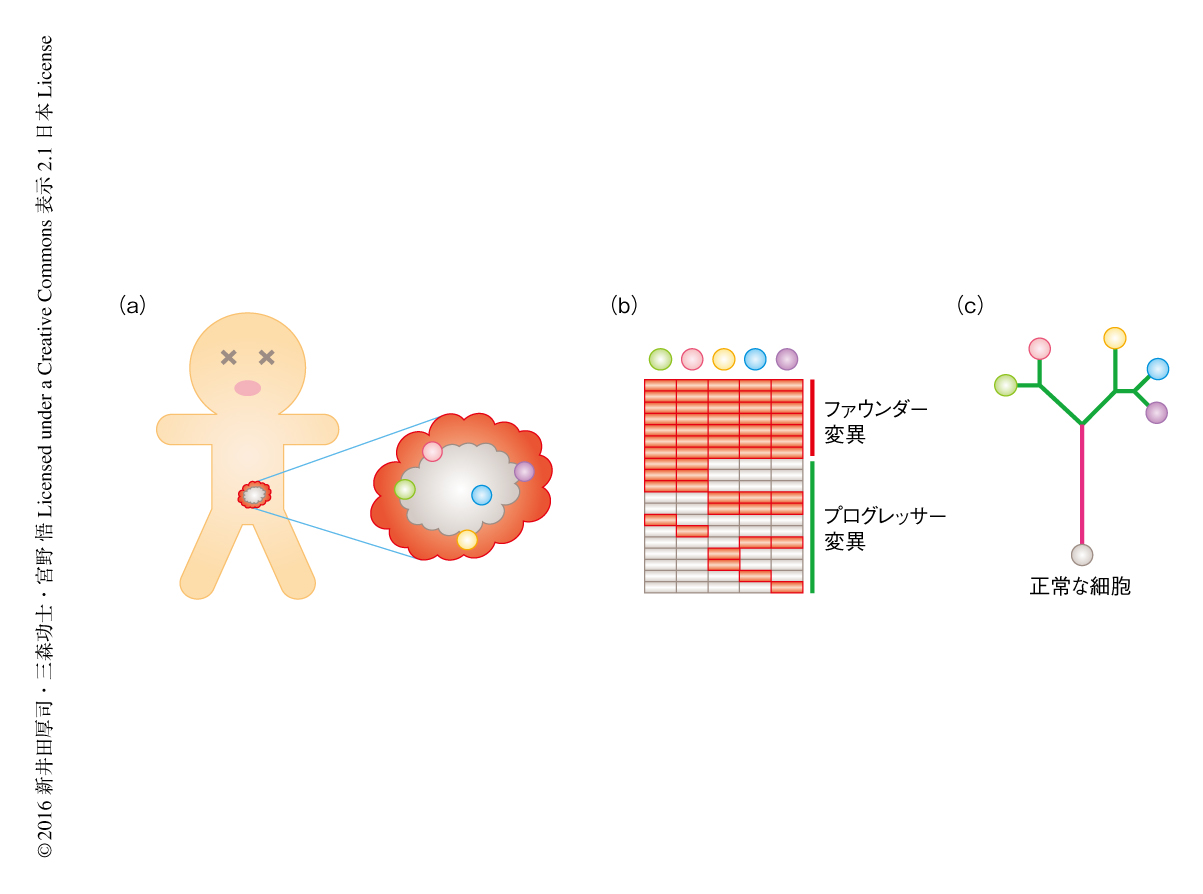
さらに,このような腫瘍間不均一性のゲノム解析にくわえ,腫瘍内不均一性についても,ここ数年の次世代シークエンサーによる包括的な解析により,複数の種類のがんにおいて,これまでの想定以上に広汎な腫瘍内不均一性の存在することが明らかにされてきた.そのような研究においては,同一の患者の腫瘍の複数の部位から得た試料において塩基配列を解読する多領域シークエンシング(multiregion sequencing)とよばれるアプローチがとられる(図1).たとえば,10症例の腎臓がんにおいて原発巣および転移巣の異なる部位から複数の検体を取得して全エキソンの塩基配列を解読した報告がある5,6).通常,多領域シークエンシングにおいては,部位に共通して存在するファウンダー変異と共通しないプログレッサー変異が観察される.クローン進化説と照らしあわせると,単一の正常な細胞がファウンダー変異を獲得して共通祖先クローンが現われ,そののち,プログレッサー変異を獲得することによりサブクローンに分岐すると考えられる.また,このような変異のパターンからは進化系統樹を推定することができる.この多領域シークエンシングにより,腎臓がんにおけるいちじるしい腫瘍内不均一性が明らかにされ,VHL遺伝子などファウンダー変異として変異を獲得する既知のドライバー遺伝子が存在する一方,SETD2遺伝子やBAP1遺伝子などのドライバー遺伝子はプログレッサー変異として変異を獲得することが明らかにされた.とくにこれらの遺伝子に関しては,症例により同一の遺伝子において平行進化により独立してプログレッサー変異を獲得することが観察された.この結果から,腫瘍内不均一性の一部は自然選択により生じることが示唆された.
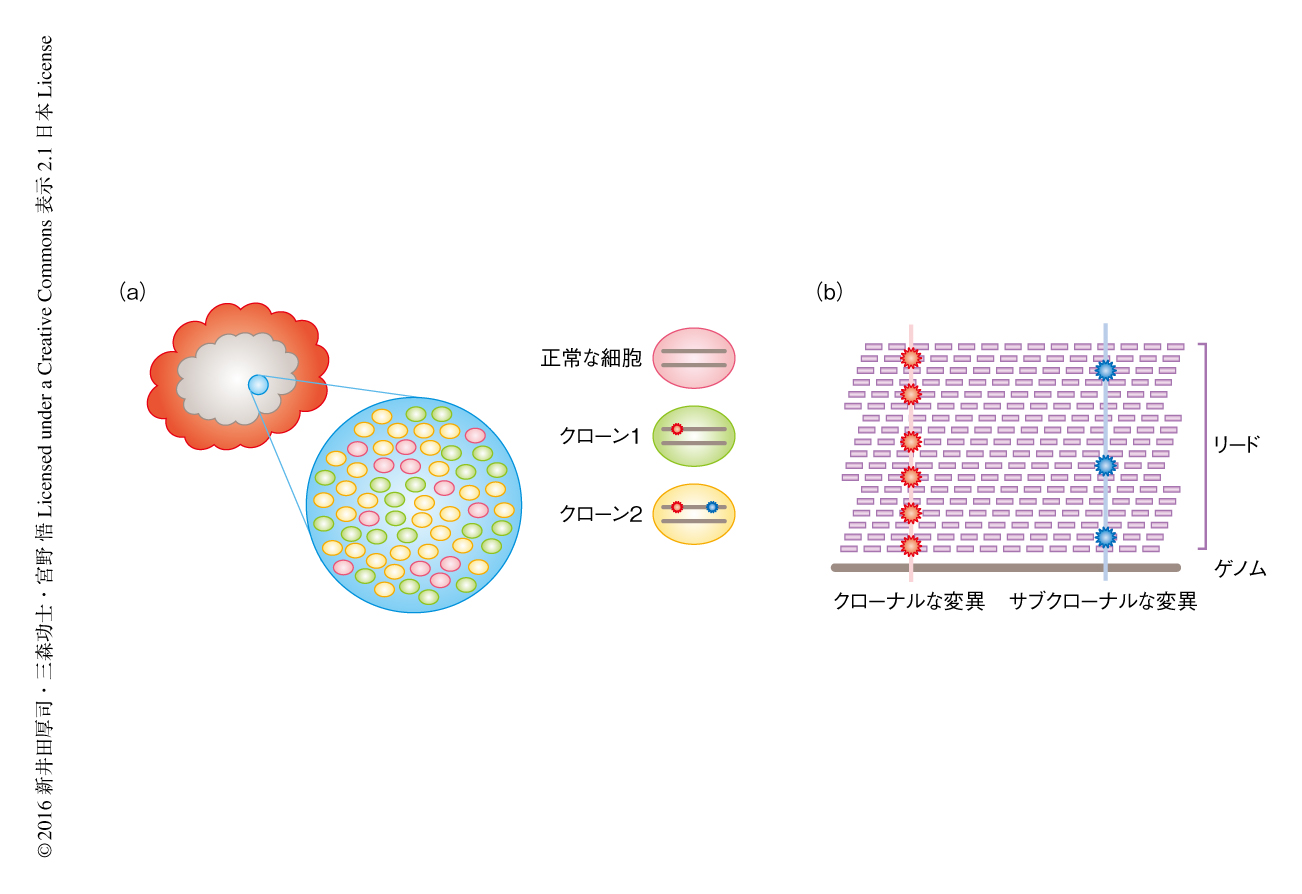
また,腫瘍の単一の部位から得た試料を高深度シークエンシング(deep sequencing)することによっても腫瘍内不均一性の情報を取得できる(図2).次世代シークエンサーの利点のひとつとして,ゲノムにおいて特定の座標の変異につき,その部位をカバーするリードを十分に取得することにより,その変異をもつアレル頻度を測定できる点があげられる.高深度シークエンシングはアレル頻度の正確な測定およびアレル頻度の低い変異の同定を目的として,リードを通常より高い数百~数千といったカバー率で解読する方法である.たとえば,21症例の乳がんにおいて原発巣から得た試料の全ゲノムについて高深度シークエンシングした報告がある7).試料のおのおのの変異のアレル頻度の情報が取得できると,それをもとにサブクローンの構造に関する情報を取得できる.もし,腫瘍が完全にクローナルでコピー数変化がなければすべてのアレル頻度は0.5になる.実際には,正常な細胞が混入しているので変異のアレル頻度は0.5より小さくなり,コピー数変化のある変異に関してもこのアレル頻度からずれることになる.さらに,サブクローンに由来する変異はコピー数変化あるいは正常な細胞の混入により説明できない,アレル頻度の低い変異として観察される.この報告では,同一のリードにおいて1塩基多型(single nucleotide polymorphism:SNP)およびサブクローナルな変異が共起しているかどうかの情報から,おのおのの変異およびコピー数変化がどのような順に起こったかを計算し,乳がんの進化の過程を推定している.このような高深度シークエンシングのデータから腫瘍内不均一性およびがんの進化の過程を推定する問題はバイオインフォマティクスの観点からも興味深く,複数の手法が開発されている8-11).
最近の研究においては,多領域シークエンシングと高深度シークエンシングのどちらかではなく,この2つのアプローチを組み合わせておのおのの部位における腫瘍内不均一性まで解析されている.たとえばこれまでに,肺がん12,13),乳がん14),前立腺がん15,16),卵巣がん17),低悪性度グリオーマ18) などにおいて,腫瘍内不均一性について報告されている.筆者らも,9症例の大腸がんの全エキソンについて多領域シークエンシングし,おのおのの症例においてファウンダー変異およびプログレッサー変異を同定した19).プログレッサー変異のパターンは腫瘍における試料の取得部位ときれいに相関しつつ,広汎な腫瘍内不均一性を示した.また,おのおのの部位においてファウンダー変異はクローナルな変異として存在するが,プログレッサー変異はサブクローナルな変異として存在することが見い出された.このことから,ひとつの部位においても多領域シークエンシングの解像度ではとらえられない不均一性の存在することが示された.また,KRAS遺伝子やAPC遺伝子など既知のドライバー遺伝子はファウンダー変異として存在する一方,腎臓がんとは対照的に,プログレッサー変異に明らかなドライバー遺伝子は少なく,平行進化も観察されなかった.ほかのがんにおいても,前立腺がんや低悪性度グリオーマなど平行進化が観察されるものがある一方,そうでないものもあり,自然選択により腫瘍内不均一性の起源のすべては説明されないことが示唆された.
ゲノム解析によりさまざまながんにおいて広汎な腫瘍内不均一性のあることが明らかにされたが,ゲノム解析のみでそれを生じる機構の理解にいたるのは困難である.そのこともあり,近年,腫瘍内不均一性を生じる進化の原理を探索することを目的とした数理的な研究が進められている.
発がんの過程に関する数理的な研究には長い歴史があり,古典的には,1971年,網膜芽細胞腫の発症年齢の解析から導き出された2ヒット説が有名である20).非遺伝性の網膜芽細胞腫にはDNAへの2つのヒットが必要である一方,遺伝性の網膜芽細胞腫を患う子どもは遺伝的に第1のヒットをもち第2のヒットのみ必要なので若年性に発症する,という仮説がたてられた.のちに,この仮説はがん抑制遺伝子としてRb遺伝子が発見されることにより証明された.
近年では,がんの進化を数理モデルで表わし進化ダイナミクスを解析する研究がさかんである.一般に,数理モデルは方程式のかたちで示され,その解を求めたり性質を調べたりされる.このアプローチは古典力学においてニュートンの運動方程式を解いて投げたボールの軌道を予測することに似ている.初期値とパラメーター,つまり,ボールの初速と質量が決定すれば軌道は正確に計算でき,放物線をかくことが予測できる.がんのシステムをボールのようにシンプルな運動方程式のかたちで記述するのはむずかしいと思われるが,理想的には,なにかしら物理的な方程式のあるものと期待される.数理的な研究においては,複雑さのなかにある本質をとらえて抽象化した方程式により表現し,システムのダイナミクスが定量的に解析される.最近のがんの数理的な研究においては,株価やブラウン運動する粒子など確率的に変動するシステムを記述するのに用いられる確率過程という理論的な枠組みがよく用いられる.多段階発がん仮説を確率モデルによりモデル化して発がんまでの時間を推定したり,直線的な進化の過程において腫瘍内不均一性のダイナミクスを追跡したりした研究が報告されている21-25).ボールの軌道についての運動方程式は解析的に求解が可能,つまり,数式を操作することにより解を求めることができる.これらの研究においても,解析的に求解の可能な方程式までがんの進化を抽象化して解析的に解かれている.
一方で,より現実なモデルは解析的に求解の可能な方程式により表現するのはむずかしいことも多い.しかしながら,そのような場合でもコンピューターを用いた数値計算により十分に正確な解を求めることができる.このアプローチはシミュレーションとよばれ,その代表例が天気予報である.雲の動きを非線形の運動方程式により記述し,観測データから得られた初期条件およびパラメーターを用いてスーパーコンピューターにより雲の動きを予測する.解析的な手法を中心とする数理的な研究は理論科学ともよばれ,長年,実験科学に相対するものとして存在してきたが,近年,計算科学ともよばれるシミュレーションを中心とする数理的な研究は,実験と理論科学とのあいだをうめる存在として重要性を増している.
がんの研究においても,シミュレーションによるがんの進化の再現が試みられている.複数の研究において用いられているシミュレーション法のひとつとして,セルオートマトン(cellular automaton)モデルがあげられる(エージェントベースドモデルの名称も使われる).セルオートマトンモデルは,セルオートマトンとよばれる独立したシステムの構成因子を仮定し,セルオートマトン自体の自立的なふるまい,セルオートマトン間およびセルオートマトン環境の相互作用の規則を規定したものである.セルオートマトンモデルはもはや数式のかたちはとらないが,シミュレーションのひとつの利点として,このような数式によらない柔軟性に富んだモデルの表現が可能な点があげられる.初期条件およびシステムのパラメーターを含むこれら規則があたえられれば複雑なシステムの挙動を簡単に再現し分析できるので,セルオートマトンモデルは社会学,経済学,生物学など分野をこえて利用されている.がんをシミュレーションする場合,おのおのの細胞をセルオートマトンとして仮定するのがもっとも自然と考えられ,腫瘍内不均一性もおのおののセルオートマトンの内部状態の違いにより容易に表現できる.たとえば,2002年には,数個の遺伝子をもつおのおのの細胞をセルオートマトンとして仮定し,それらに変異を導入しながら増殖させることにより,実験データにみられるような空間的な腫瘍不均一性が再現されている26).それ以降も,セルオートマトンモデルを用いたがんの進化シミュレーションにより腫瘍不均一性の生じる機構を探索する複数の研究が報告されている.がん幹細胞の存在がゲノムの腫瘍内不均一性に寄与する可能性が示され27),3次元の空間における細胞の相互作用およびターンオーバーが腫瘍内不均一性にあたえる影響が解析された28).
しかしながら,最近のゲノム解析により明らかにされた広汎な腫瘍内不均一性を十分に表現できるモデルは構築されていない.筆者らは,大腸がんにおける多領域シークエンシングの結果を再現し腫瘍内不均一性を生じる原理を探索することを目的として,新規のセルオートマトンモデルとしてBEP(branching evolutionary processs)モデルを構築した19,29)(図3a).このBEPモデルにおいても,おのおのの細胞をセルオートマトンとして仮定する.おのおのの細胞はd個のドライバー遺伝子を含むn個の遺伝子をもち,単位時間あたり確率pで分裂し確率qで死ぬとする.さらに,細胞が分裂する際,おのおのの遺伝子には変異率rでランダムに変異が導入され,ドライバー遺伝子が変異を獲得すると増殖速度pが10f倍ずつ増加する.つまり,fはドライバー遺伝子の強さとみなせる.適当なパラメーター値をあたえ以上の規則にしたがい細胞を増殖させていくと,変異のない正常な細胞が増殖していく過程においてひとつの細胞がひとつのドライバー遺伝子の変異を獲得する.すると,そのクローンの増殖速度が上昇し,自然選択をうけ細胞の集団における割合が上昇する.このようなステップをくり返すことによりおのおのの細胞は確率的にドライバー遺伝子の変異,および,それに付随する増殖速度に影響をあたえないパッセンジャー遺伝子の変異を蓄積し,最終的に,多くの変異を蓄積したがん細胞があつまった腫瘍が形成される.パラメーターの値にもよるが,この進化の過程においておのおののがん細胞に異なる組合せの変異が蓄積され腫瘍内不均一性が形成される.例として,適当なパラメーター値でBEPモデルによるシミュレーションを行い,2次元の空間でがんを成長させたようすを示す(図3 b, c).この例では,正常な細胞に順にドライバー遺伝子の変異が蓄積し,最終的に4つのドライバー遺伝子の変異をもつクローンが自然選択をうけ多数をしめた.
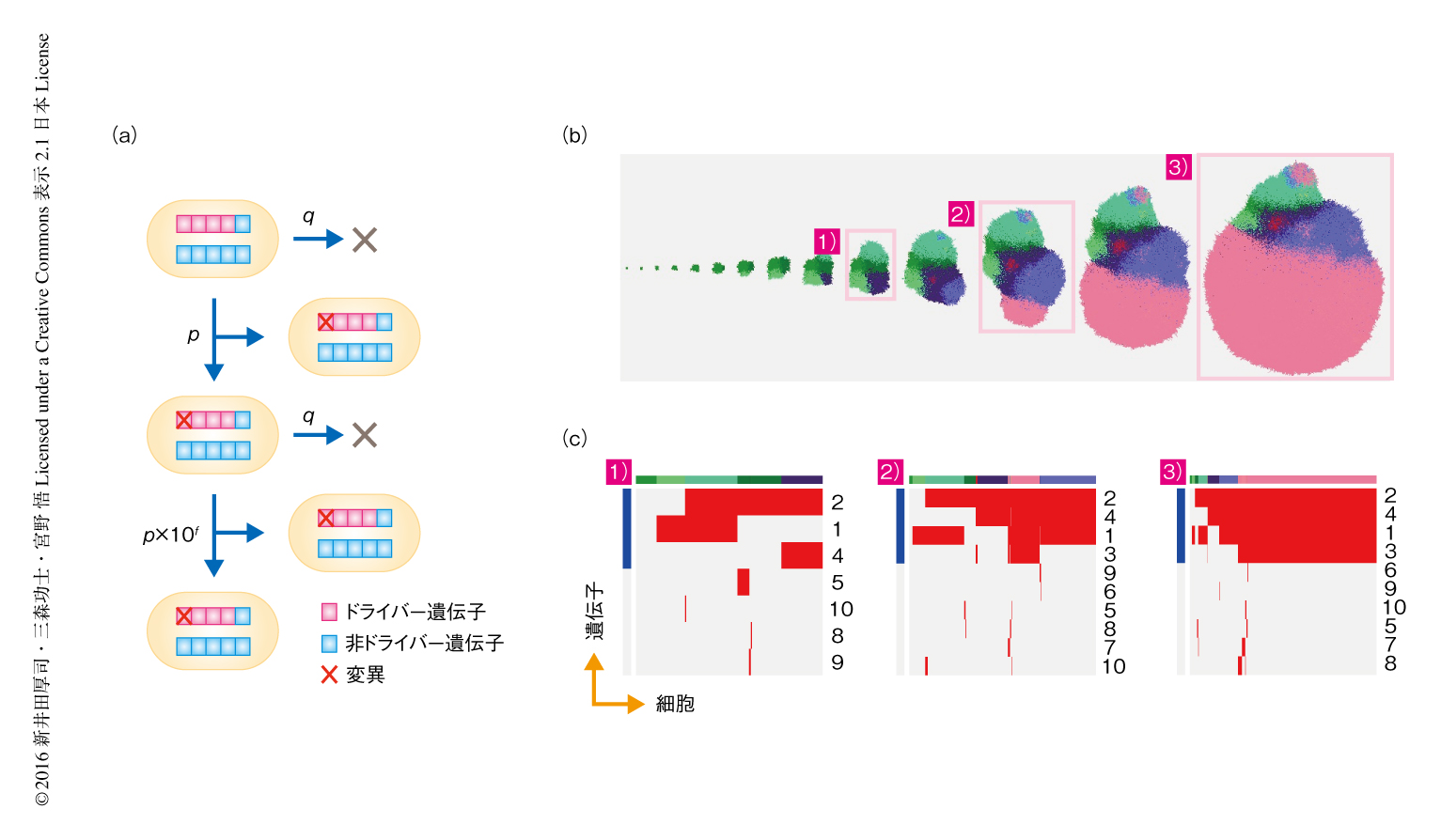
腫瘍内不均一性の生じる原理を探索するため,スーパーコンピューターによりさまざまなパラメーター値でBEPモデルによるシミュレーションを行うことにより,ゲノム解析から観察される広汎な腫瘍内不均一性が生じる条件を探索した.その結果,高い遺伝子変異率を仮定してがんの進化をシミュレーションし,さらに,in silicoにおいて形成されたがんを多領域シークエンシングしたところ,大腸がんにおける多領域シークエンシングから得られた変異プロファイルと類似した結果が得られた.すなわち,ファウンダー変異が存在する一方でプログレッサー変異が試料の取得部位と相関した腫瘍内不均一性を示し,かつ,おのおのの部位ごとにプログレッサー変異はサブクローナルな変異として存在し局所における不均一性を示した.さらに,ドライバー遺伝子の変異はファウンダー変異として獲得された一方,プログレッサー変異にドライバー遺伝子の変異はほとんど含まれず,大部分は細胞の増殖速度に影響をあたえない中立の変異であった.つまりこのことから,自然選択によりドライバー遺伝子の変異が蓄積された共通祖先クローンが形成されたのち,中立進化により広汎な腫瘍内不均一性が生じた可能性が示された.中立進化とは,ダーウィンにより提唱された古典的な自然選択による進化に対し,有利でもなく不利でもない中立な変異が集団に偶然に広まったことにより起こる進化の形式であり,1968年,木村資生により提唱された30).発表された当時,中立進化説は古典的な自然選択説の支持者による激しい抵抗にあったが,現在では,生物種間の遺伝的な多様性はおもに中立進化により生じるということは広く受け入れられている.BEPモデルによるシミュレーションにてこのような中立進化が起こる条件においては,ひとつの細胞の変異プロファイルより多領域シークエンシングの解像度ではとらえきれない無数の中立変異を蓄積する微小なクローンが存在するであろうことも予測された.また,高い変異率の代わりにモデルを拡張してがん幹細胞の存在を仮定しても,同様に中立進化の起こりうることが見い出された.
最近,ほかの研究グループからも,筆者らと同様な結果が報告されている.大腸がんにおける腫瘍内不均一性を多領域シークエンシングと高深度シークエンシングを組み合わせて解析することにより,腫瘍の全体にわたる均一な腫瘍内不均一性が見い出された31).さらに,腫瘍のある部位に検出されたプログレッサー変異がそこからはなれた部位にアレル頻度の低いサブクローナルな変異として混ざり込む現象に注目しシミュレーションを用いて検証することにより,ビッグバンモデルが提唱された.このビッグバンモデルでは,がんの進化の初期に多くのサブクローンが生じ,そののち,それらが一部混ざり合いながら自然選択をうけずに広がり,腫瘍のどの部分においても一様に高い不均一性が形成されるとする.また,肝細胞がんにおける多領域シークエンシングのデータから得られたクローンの多様性に関する統計量の計測値が,解析的に導かれた中立進化が成り立つときに得られる理論値と統計的に一致することが示され,腫瘍内不均一性は中立進化により生じると主張された32,33).中立進化により無数の変異がひとつの腫瘍に蓄積されているとも主張され,これは筆者らの主張とも一致した.
ここまで,腫瘍内不均一性を理解するためのがんのゲノム解析および数理的な研究につき,最近の研究について解説した.ゲノム解析により種々のがんにおける広汎な腫瘍内不均一性の存在が示され,数理的な研究からは中立進化により腫瘍内不均一性が生じる可能性が示された.ここから,今後,期待される展開について述べる.
このレビューにおいては,ゲノム,とくに1塩基変異にしぼり腫瘍内不均一性について述べてきたが,オミックスのほかの階層においても同様な腫瘍内不均一性のあるものと思われる.筆者らも,大腸がんにおいて1塩基変異にくわえ,DNAコピー数,DNAメチル化,トランスクリプトームの腫瘍内不均一性が互いに相関しあいながら存在することを確認している19).ほかの研究グループからも,前立腺がんにおけるDNAメチル化の腫瘍内不均一性について報告されている34).また,最近,開発の進んでいる1細胞シークエンシングの技術を利用すれば,オミックスのおのおのの階層における腫瘍内不均一性を究極の解像度でみることが可能になるだろう35).たとえば,乳がんにおいてゲノムの1細胞シークエンシングが報告されている36).ゲノムの1細胞シークエンシングはDNAコピー数に関しては信頼性のあるデータが得られるようになっているものの,1塩基変異に関しては技術的なノイズの問題があり完全な1細胞の変異プロファイルを得るのはまだむずかしいのが現状である.そこで,複数の細胞において同定された1塩基変異の高深度シークエンシングにより,多くの変異が低い頻度で存在することが確認された.トランスクリプトームに関しては腫瘍からの試料の調製のむずかしさという問題はあるものの,1細胞RNA-seq法それ自体は技術的にほぼ完成されている.たとえば,グリオブラストーマにおいてトランスクリプトームの広汎な腫瘍内不均一性が1細胞RNA-seq法により明らかにされている37).DNAメチル化に関しても1細胞バイサルファイトシークエンシング法の開発が進んでおり,今後のがんの研究への応用が期待される38).
このような新しい実験技術の登場により,今後とも,腫瘍内不均一性に関するデータは量質ともにますます増えていくだろう.データが増加するにつれて,膨大なデータのなかの本質をとらえる数理的な研究の重要性が増してくる.これまでに得られているデータのなかからでも解かれるべき問題がある.たとえば,筆者らにより,大腸がんにおいて平行進化は観察されず腫瘍内不均一性は中立進化により説明される可能性のあることが示された一方で,腎臓がんおよびほかのがん腫においては平行進化がみられ,がんによっては腫瘍内不均一性の一部は自然選択により生じている可能性もある.筆者らは,BEPモデルによるシミュレーションにおいて,中立進化による無数のサブクローンをもつ腫瘍にくわえ,変異率が低く強いドライバー遺伝子のあるときには自然選択により均一な腫瘍が,変異率が低く弱いドライバー遺伝子が複数あるときには自然選択により複数のサブクローンをもつ腫瘍が生じることを見い出した.つまり,H2Oが温度や気圧というパラメーターの違いより氷,水,水蒸気の異なる相をとるように,がんもドライバー遺伝子の数,強さ,変異率などのパラメーターの違いにより異なる進化ダイナミクスの相を示すと考えられた.これらのパラメーターががんにより異なるのはもっともだと思われるし,同じがんの進化の過程においても変異率などは変化していくと想像される.今後,BEPモデルあるいはより抽象化した数理モデルを解析的に分析したり,種々のがんについてパラメーターのデータを収集したりすることにより,腫瘍内不均一性の生じる原理の詳細をさらにつめていく必要がある.
近年の計算資源の拡大とともに,とくにシミュレーションを中心とする計算科学の可能性が拡大している.筆者らの研究も,スーパーコンピューターを利用することによりはじめて可能となった.BEPモデルによるシミュレーションは確率的であり結果が試行ごとに異なるため,同じ条件の複数の試行の平均をみる必要があり,さらに,十分なパラメーター空間を探索するとなると膨大な計算時間を必要とするため通常の計算資源ではむずかしい.また.生物システムのシミュレーションのむずかしさのひとつとして,多くの内部パラメーターの値が実験的に直接には測定できず正確には決まっていないことがあげられる.そのような場合,観測データからパラメーター値を推定する必要があるが,そのようなパラメーターの推定の方法として,近年,実データとシミュレーションモデルがあたえられたときに実データを生じるパラメーター値の確率分布を計算できるapproximate Bayesian computation(ABC)法が注目されている39).もっとも簡単なかたちのABC法では,おのおののパラメーター値(複数のパラメーターがある場合にはその組合せ)について複数回のシミュレーションデータを生成し,データの特徴を記述する要約統計量を用いてシミュレーションデータと実データの類似性を評価して,おのおののパラメーター値において類似したシミュレーションデータが得られる確率を計算する.ベイズの定理より原因と結果を逆転させることができ,この確率は実データがあたえられたときにそのようなデータを生じることのできるパラメーター値の確率分布になる.筆者らも,BEPモデルによるシミュレーションにおいて,実データと同様の変異プロファイルを生じるパラメーター値の探索にABC法を用いた19).さらに,ABC法はパラメーターの推定だけでなくモデルの選択にも適用が可能である.つまり,ABC法を用いてあたえられたデータをよりよく再現できるようモデルを改良することができる.筆者らによるBEPモデルはまだトイモデルの域をでていないが,今後,ますます増えていくと思われる腫瘍内不均一性に関する実験データをABC法のようなデータ同化手法を用いて数理モデルに取り込んでいくことにより,より現実的ながんの進化のシミュレーションが行えるであろう.
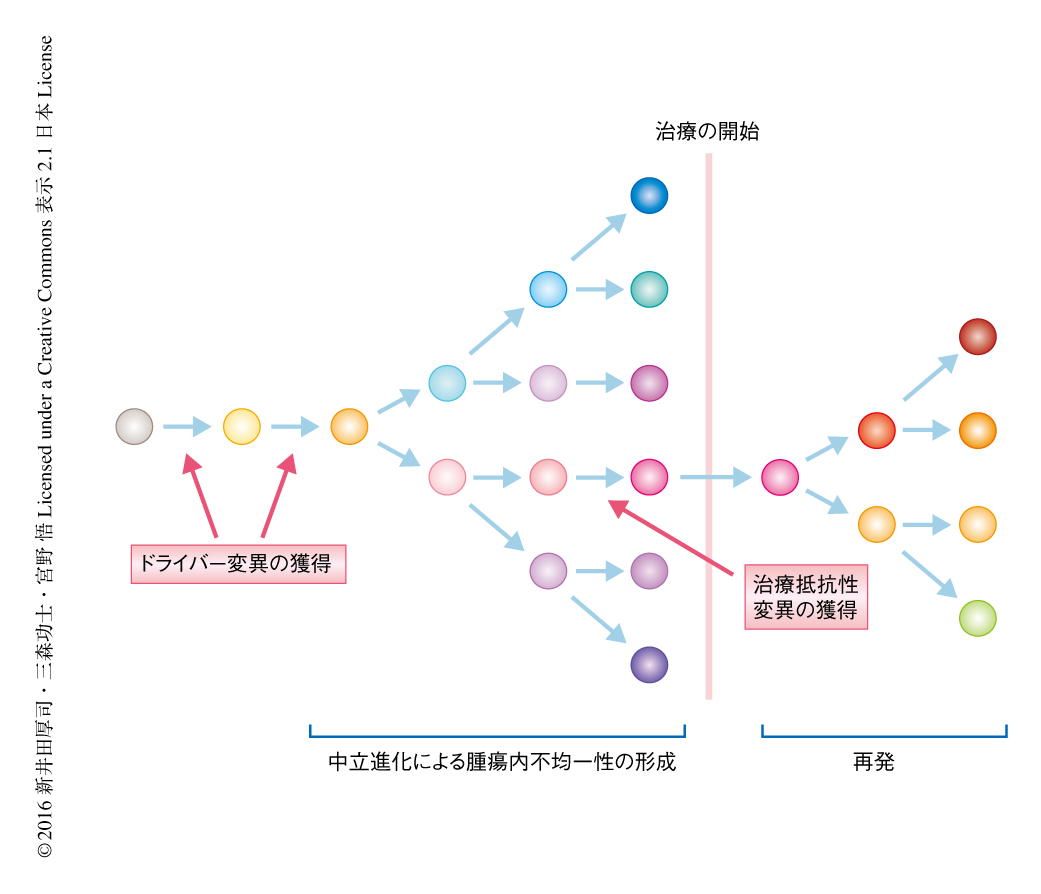
がんの研究分野においては,20世紀後半の分子生物学の進展により発がんの分子機構が明らかにされ,現在,その知見をもとにさまざまな分子標的薬が開発されている.また,次世代シークエンサーを利用しさまざまながんにおいて変異のカタログ化が進行しており,今後,臨床における塩基配列の解読と分子標的薬とを組み合わせた個別化医療が進んでいくと考えられる.しかしながら,多くのがんにおいて治療抵抗性の問題は解決されておらず,治療により一時的に縮小した腫瘍も,多くの場合,結局は再発してしまう.筆者らによるシミュレーションの結果より,ひとつの腫瘍のなかに中立進化により生じた無数のクローンが存在していることが予測されたが,この結果は,がんの治療抵抗性の根本的な原因である可能性がある.中立な変異であるかどうかは環境にも依存するので,治療のまえには中立な変異であっても,治療により環境が変わればドライバー遺伝子の変異,つまり,治療抵抗性の変異になりうる.すなわち,どのような治療をしても無数のクローンのなかにそのような変異を獲得した治療抵抗性のクローンが存在してしまい,結局は再発にいたることが考えられる(図4).最近の大腸がんにおける抗EGFR抗体医薬の治療耐性の獲得に関する研究も,血中マーカーのダイナミクスの数理解析により,治療の開始の際にすでに治療抵抗性のクローンは存在しているという見方が支持されている40).
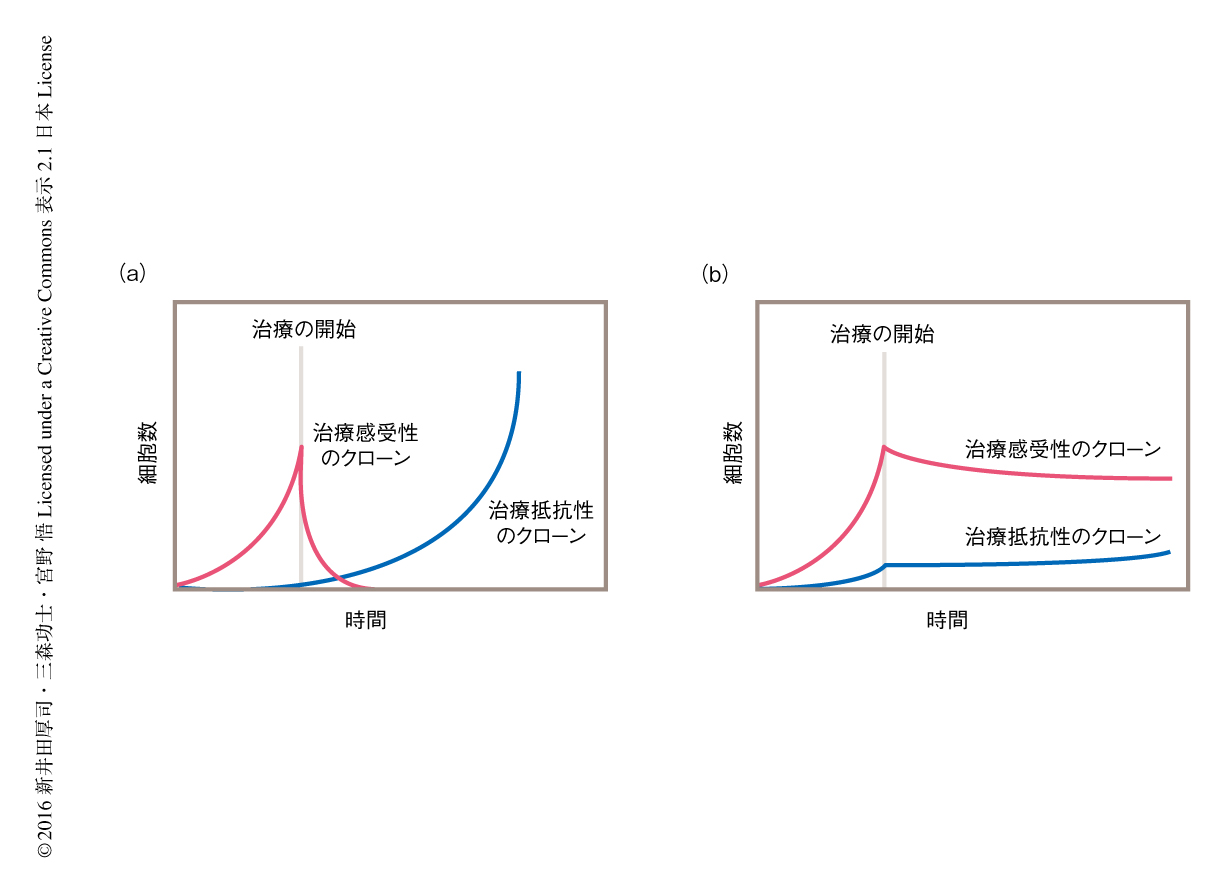
近年,治療抵抗性および感受性クローンを仮定したモデルを用いた数理的な研究により,抗がん剤の投薬の計画を調整することにより,がんの再発を遅延あるいは阻止させることのできる可能性が示されている41,42)(図5).たとえば,ひとつの腫瘍に多数の治療感受性のクローンと少数の治療抵抗性のクローンが存在する場合,通常のように大用量の抗がん剤を継続的に投与すれば治療感受性のクローンが減少することにより腫瘍は一時的に縮小するが,やがて,治療抵抗性クローンが増殖することによりがんは再発にいたる.一方で,もし用量を調整して治療感受性クローンをすべて殺さずに治療抵抗性クローンとうまく競合させることができれば,全体としてがんの成長を遅らせて患者の予後を伸ばすことが可能と思われる.また,HIVの治療において行われているように,複数の薬剤を使うことにより治療抵抗性のクローンの出現を抑えられることも示されている43).がんの治療において治療抵抗性の克服は最重要課題のひとつであるが,このような試みに腫瘍内不均一性の解析の研究から見い出された中立進化のモデルを組み込むことにより,解決の糸口がみえてくる可能性がある.
これまで,がんの研究分野は分子生物学など個別的あるいは定性的な実験科学が主流であり,一部の例外をのぞき,理論科学および計算科学は華やかな実験科学の陰にかくれていた感がある.しかしながら,現在,次世代シークエンサーに代表される新技術の登場により包括的かつ定量的なデータの取得が可能になり,ようやく,理論科学および計算科学が表舞台に浮上し実験科学と融合する下地が整いつつある.がんの治療は日々進歩しているものの,がんはいまだ不治の病であることに変わりはない.筆者らは,がんを根本的に克服するためには異分野との融合研究によるがんの進化のシステムレベルでの原理的な理解が必須であると考えている.今後,さまざまな分野から研究者が参入し,がんの研究における異分野との融合が進むことを期待しつつ筆をおく.
略歴:2007年 東京大学大学院理学系研究科 修了,同年 東京大学分子細胞生物学研究所 研究員,2008年 東京大学医科学研究所 研究員を経て,2011年より同 特任助教(現 助教).
研究テーマ:がんのバイオインフォマティクスおよび数理解析.
抱負:実験と数理,および,基礎と臨床の共同研究をつうじて,がんの進化の原理の解明,治療抵抗性の克服,がんの進化のシミュレーションの臨床応用をめざしている.
三森 功士(Koshi Mimori)
九州大学病院別府病院 教授.
宮野 悟(Satoru Miyano)
東京大学医科学研究所 教授.
© 2016 新井田厚司・三森功士・宮野 悟 Licensed under CC 表示 2.1 日本
(1東京大学医科学研究所 ヘルスインテリジェンスセンター健康医療計算科学分野,2九州大学病院別府病院 外科)
email:新井田厚司
領域融合レビュー, 5, e003 (2016) DOI: 10.7875/leading.author.5.e003
Atsushi Niida, Koshi Mimori & Satoru Miyano: Intratumor heterogeneity and cancer evolution.
要 約
長年,がんは直線的なクローン進化をへて形成される悪性度の高いクローナルな細胞の集団としてとらえられてきた.しかしながら,近年,次世代シークエンサーを用いたゲノム解析により,ひとつの腫瘍のなかにも多数のサブクローンの存在することが明らかにされている.さらに,がんの進化のシミュレーションなどにより,この腫瘍内不均一性は中立進化により生じる可能性が示されている.腫瘍内不均一性はがんの治療抵抗性の根本的な原因である可能性もあり,腫瘍内不均一性を生じるがんの進化のシステムレベルでの原理的な理解は臨床的にも重要である.そのためには,今後,がんの研究において異分野との融合を推進していく必要がある.それをみすえて,このレビューにおいては,この分野のこれまでの知見および期待される展開について解説する.
はじめに
ひとつの腫瘍のなかにはゲノムの異なる複数のクローンの存在することが知られており,この現象は腫瘍内不均一性とよばれている.腫瘍内不均一性はさまざまなタイプのがんにおいて観察されており,がんの治療抵抗性の一因になっていると考えられる.つまり,腫瘍内不均一性があると,治療感受性のクローンが大多数をしめる腫瘍が治療により縮小しても,治療抵抗性の少数のクローンが残存する可能性があり,やがて,そのようなクローンが増殖して再発にいたることが考えられる.腫瘍内不均一性はがんの進化の過程でクローンが分岐することにより生じると考えられる.
1976年,体細胞変異の獲得および変異を獲得したクローンへの自然選択が段階的に起こることにより,がんは単一の正常な細胞から生じることが提唱された1).つまり,がん細胞の進化は非生殖分裂をする単細胞生物のダーウィン的な進化と同様にとらえることができる.このがんのクローン進化説が提唱されたのち,分子生物学によりがん遺伝子およびがん抑制遺伝子が発見され,1990年,これらの発見とクローン進化説は多段階発がん仮説として統合された2).すなわち,大腸がんの発がんの過程において,APC遺伝子,KRAS遺伝子,TP53遺伝子などに発がんの原因となる複数の変異が順に蓄積することにより,正常な上皮細胞が良性腫瘍をへて悪性腫瘍へと直線的に変化するというモデルが提唱された.それ以降,個別の遺伝子レベルでの実験において腫瘍内不均一性の存在が示唆されてきたものの,直線的なクローンの進化をへて悪性度の高いクローナルな細胞の集団として腫瘍が形成されるというモデルが一般的な発がんの過程のイメージとして受け入れられてきた.
1.ゲノム解析による腫瘍内不均一性の解明
近年,次世代シークエンサー(next generation sequencer:NGS)の登場により,がんにおける体細胞変異の包括的なプロファイル,すなわち,がんゲノムを安価かつ迅速に解読することが可能になり,これまで分子生物学が中心であったがんの研究分野において急速なパラダイムシフトが起こっている.The Cancer Genome Atlas(TCGA)やInternational Cancer Genome Consortium(ICGC)におけるような多数の患者から得た試料において塩基配列を解読するゲノム研究により,その変異ががんの発生や進展の直接的な原因になるドライバー遺伝子が数多く同定され,さまざまながんにおいて変異のカタログが作成されている.また,これらの研究により,同一のがんの患者のあいだでもがんゲノムにいちじるしい不均一性のあることが明らかにされ,個別化医療の必要性がとなえられている3,4).
さらに,このような腫瘍間不均一性のゲノム解析にくわえ,腫瘍内不均一性についても,ここ数年の次世代シークエンサーによる包括的な解析により,複数の種類のがんにおいて,これまでの想定以上に広汎な腫瘍内不均一性の存在することが明らかにされてきた.そのような研究においては,同一の患者の腫瘍の複数の部位から得た試料において塩基配列を解読する多領域シークエンシング(multiregion sequencing)とよばれるアプローチがとられる(図1).たとえば,10症例の腎臓がんにおいて原発巣および転移巣の異なる部位から複数の検体を取得して全エキソンの塩基配列を解読した報告がある5,6).通常,多領域シークエンシングにおいては,部位に共通して存在するファウンダー変異と共通しないプログレッサー変異が観察される.クローン進化説と照らしあわせると,単一の正常な細胞がファウンダー変異を獲得して共通祖先クローンが現われ,そののち,プログレッサー変異を獲得することによりサブクローンに分岐すると考えられる.また,このような変異のパターンからは進化系統樹を推定することができる.この多領域シークエンシングにより,腎臓がんにおけるいちじるしい腫瘍内不均一性が明らかにされ,VHL遺伝子などファウンダー変異として変異を獲得する既知のドライバー遺伝子が存在する一方,SETD2遺伝子やBAP1遺伝子などのドライバー遺伝子はプログレッサー変異として変異を獲得することが明らかにされた.とくにこれらの遺伝子に関しては,症例により同一の遺伝子において平行進化により独立してプログレッサー変異を獲得することが観察された.この結果から,腫瘍内不均一性の一部は自然選択により生じることが示唆された.
また,腫瘍の単一の部位から得た試料を高深度シークエンシング(deep sequencing)することによっても腫瘍内不均一性の情報を取得できる(図2).次世代シークエンサーの利点のひとつとして,ゲノムにおいて特定の座標の変異につき,その部位をカバーするリードを十分に取得することにより,その変異をもつアレル頻度を測定できる点があげられる.高深度シークエンシングはアレル頻度の正確な測定およびアレル頻度の低い変異の同定を目的として,リードを通常より高い数百~数千といったカバー率で解読する方法である.たとえば,21症例の乳がんにおいて原発巣から得た試料の全ゲノムについて高深度シークエンシングした報告がある7).試料のおのおのの変異のアレル頻度の情報が取得できると,それをもとにサブクローンの構造に関する情報を取得できる.もし,腫瘍が完全にクローナルでコピー数変化がなければすべてのアレル頻度は0.5になる.実際には,正常な細胞が混入しているので変異のアレル頻度は0.5より小さくなり,コピー数変化のある変異に関してもこのアレル頻度からずれることになる.さらに,サブクローンに由来する変異はコピー数変化あるいは正常な細胞の混入により説明できない,アレル頻度の低い変異として観察される.この報告では,同一のリードにおいて1塩基多型(single nucleotide polymorphism:SNP)およびサブクローナルな変異が共起しているかどうかの情報から,おのおのの変異およびコピー数変化がどのような順に起こったかを計算し,乳がんの進化の過程を推定している.このような高深度シークエンシングのデータから腫瘍内不均一性およびがんの進化の過程を推定する問題はバイオインフォマティクスの観点からも興味深く,複数の手法が開発されている8-11).
最近の研究においては,多領域シークエンシングと高深度シークエンシングのどちらかではなく,この2つのアプローチを組み合わせておのおのの部位における腫瘍内不均一性まで解析されている.たとえばこれまでに,肺がん12,13),乳がん14),前立腺がん15,16),卵巣がん17),低悪性度グリオーマ18) などにおいて,腫瘍内不均一性について報告されている.筆者らも,9症例の大腸がんの全エキソンについて多領域シークエンシングし,おのおのの症例においてファウンダー変異およびプログレッサー変異を同定した19).プログレッサー変異のパターンは腫瘍における試料の取得部位ときれいに相関しつつ,広汎な腫瘍内不均一性を示した.また,おのおのの部位においてファウンダー変異はクローナルな変異として存在するが,プログレッサー変異はサブクローナルな変異として存在することが見い出された.このことから,ひとつの部位においても多領域シークエンシングの解像度ではとらえられない不均一性の存在することが示された.また,KRAS遺伝子やAPC遺伝子など既知のドライバー遺伝子はファウンダー変異として存在する一方,腎臓がんとは対照的に,プログレッサー変異に明らかなドライバー遺伝子は少なく,平行進化も観察されなかった.ほかのがんにおいても,前立腺がんや低悪性度グリオーマなど平行進化が観察されるものがある一方,そうでないものもあり,自然選択により腫瘍内不均一性の起源のすべては説明されないことが示唆された.
2.腫瘍内不均一性を生じる進化の原理を探索する数理的な研究
ゲノム解析によりさまざまながんにおいて広汎な腫瘍内不均一性のあることが明らかにされたが,ゲノム解析のみでそれを生じる機構の理解にいたるのは困難である.そのこともあり,近年,腫瘍内不均一性を生じる進化の原理を探索することを目的とした数理的な研究が進められている.
発がんの過程に関する数理的な研究には長い歴史があり,古典的には,1971年,網膜芽細胞腫の発症年齢の解析から導き出された2ヒット説が有名である20).非遺伝性の網膜芽細胞腫にはDNAへの2つのヒットが必要である一方,遺伝性の網膜芽細胞腫を患う子どもは遺伝的に第1のヒットをもち第2のヒットのみ必要なので若年性に発症する,という仮説がたてられた.のちに,この仮説はがん抑制遺伝子としてRb遺伝子が発見されることにより証明された.
近年では,がんの進化を数理モデルで表わし進化ダイナミクスを解析する研究がさかんである.一般に,数理モデルは方程式のかたちで示され,その解を求めたり性質を調べたりされる.このアプローチは古典力学においてニュートンの運動方程式を解いて投げたボールの軌道を予測することに似ている.初期値とパラメーター,つまり,ボールの初速と質量が決定すれば軌道は正確に計算でき,放物線をかくことが予測できる.がんのシステムをボールのようにシンプルな運動方程式のかたちで記述するのはむずかしいと思われるが,理想的には,なにかしら物理的な方程式のあるものと期待される.数理的な研究においては,複雑さのなかにある本質をとらえて抽象化した方程式により表現し,システムのダイナミクスが定量的に解析される.最近のがんの数理的な研究においては,株価やブラウン運動する粒子など確率的に変動するシステムを記述するのに用いられる確率過程という理論的な枠組みがよく用いられる.多段階発がん仮説を確率モデルによりモデル化して発がんまでの時間を推定したり,直線的な進化の過程において腫瘍内不均一性のダイナミクスを追跡したりした研究が報告されている21-25).ボールの軌道についての運動方程式は解析的に求解が可能,つまり,数式を操作することにより解を求めることができる.これらの研究においても,解析的に求解の可能な方程式までがんの進化を抽象化して解析的に解かれている.
一方で,より現実なモデルは解析的に求解の可能な方程式により表現するのはむずかしいことも多い.しかしながら,そのような場合でもコンピューターを用いた数値計算により十分に正確な解を求めることができる.このアプローチはシミュレーションとよばれ,その代表例が天気予報である.雲の動きを非線形の運動方程式により記述し,観測データから得られた初期条件およびパラメーターを用いてスーパーコンピューターにより雲の動きを予測する.解析的な手法を中心とする数理的な研究は理論科学ともよばれ,長年,実験科学に相対するものとして存在してきたが,近年,計算科学ともよばれるシミュレーションを中心とする数理的な研究は,実験と理論科学とのあいだをうめる存在として重要性を増している.
がんの研究においても,シミュレーションによるがんの進化の再現が試みられている.複数の研究において用いられているシミュレーション法のひとつとして,セルオートマトン(cellular automaton)モデルがあげられる(エージェントベースドモデルの名称も使われる).セルオートマトンモデルは,セルオートマトンとよばれる独立したシステムの構成因子を仮定し,セルオートマトン自体の自立的なふるまい,セルオートマトン間およびセルオートマトン環境の相互作用の規則を規定したものである.セルオートマトンモデルはもはや数式のかたちはとらないが,シミュレーションのひとつの利点として,このような数式によらない柔軟性に富んだモデルの表現が可能な点があげられる.初期条件およびシステムのパラメーターを含むこれら規則があたえられれば複雑なシステムの挙動を簡単に再現し分析できるので,セルオートマトンモデルは社会学,経済学,生物学など分野をこえて利用されている.がんをシミュレーションする場合,おのおのの細胞をセルオートマトンとして仮定するのがもっとも自然と考えられ,腫瘍内不均一性もおのおののセルオートマトンの内部状態の違いにより容易に表現できる.たとえば,2002年には,数個の遺伝子をもつおのおのの細胞をセルオートマトンとして仮定し,それらに変異を導入しながら増殖させることにより,実験データにみられるような空間的な腫瘍不均一性が再現されている26).それ以降も,セルオートマトンモデルを用いたがんの進化シミュレーションにより腫瘍不均一性の生じる機構を探索する複数の研究が報告されている.がん幹細胞の存在がゲノムの腫瘍内不均一性に寄与する可能性が示され27),3次元の空間における細胞の相互作用およびターンオーバーが腫瘍内不均一性にあたえる影響が解析された28).
しかしながら,最近のゲノム解析により明らかにされた広汎な腫瘍内不均一性を十分に表現できるモデルは構築されていない.筆者らは,大腸がんにおける多領域シークエンシングの結果を再現し腫瘍内不均一性を生じる原理を探索することを目的として,新規のセルオートマトンモデルとしてBEP(branching evolutionary processs)モデルを構築した19,29)(図3a).このBEPモデルにおいても,おのおのの細胞をセルオートマトンとして仮定する.おのおのの細胞はd個のドライバー遺伝子を含むn個の遺伝子をもち,単位時間あたり確率pで分裂し確率qで死ぬとする.さらに,細胞が分裂する際,おのおのの遺伝子には変異率rでランダムに変異が導入され,ドライバー遺伝子が変異を獲得すると増殖速度pが10f倍ずつ増加する.つまり,fはドライバー遺伝子の強さとみなせる.適当なパラメーター値をあたえ以上の規則にしたがい細胞を増殖させていくと,変異のない正常な細胞が増殖していく過程においてひとつの細胞がひとつのドライバー遺伝子の変異を獲得する.すると,そのクローンの増殖速度が上昇し,自然選択をうけ細胞の集団における割合が上昇する.このようなステップをくり返すことによりおのおのの細胞は確率的にドライバー遺伝子の変異,および,それに付随する増殖速度に影響をあたえないパッセンジャー遺伝子の変異を蓄積し,最終的に,多くの変異を蓄積したがん細胞があつまった腫瘍が形成される.パラメーターの値にもよるが,この進化の過程においておのおののがん細胞に異なる組合せの変異が蓄積され腫瘍内不均一性が形成される.例として,適当なパラメーター値でBEPモデルによるシミュレーションを行い,2次元の空間でがんを成長させたようすを示す(図3 b, c).この例では,正常な細胞に順にドライバー遺伝子の変異が蓄積し,最終的に4つのドライバー遺伝子の変異をもつクローンが自然選択をうけ多数をしめた.
腫瘍内不均一性の生じる原理を探索するため,スーパーコンピューターによりさまざまなパラメーター値でBEPモデルによるシミュレーションを行うことにより,ゲノム解析から観察される広汎な腫瘍内不均一性が生じる条件を探索した.その結果,高い遺伝子変異率を仮定してがんの進化をシミュレーションし,さらに,in silicoにおいて形成されたがんを多領域シークエンシングしたところ,大腸がんにおける多領域シークエンシングから得られた変異プロファイルと類似した結果が得られた.すなわち,ファウンダー変異が存在する一方でプログレッサー変異が試料の取得部位と相関した腫瘍内不均一性を示し,かつ,おのおのの部位ごとにプログレッサー変異はサブクローナルな変異として存在し局所における不均一性を示した.さらに,ドライバー遺伝子の変異はファウンダー変異として獲得された一方,プログレッサー変異にドライバー遺伝子の変異はほとんど含まれず,大部分は細胞の増殖速度に影響をあたえない中立の変異であった.つまりこのことから,自然選択によりドライバー遺伝子の変異が蓄積された共通祖先クローンが形成されたのち,中立進化により広汎な腫瘍内不均一性が生じた可能性が示された.中立進化とは,ダーウィンにより提唱された古典的な自然選択による進化に対し,有利でもなく不利でもない中立な変異が集団に偶然に広まったことにより起こる進化の形式であり,1968年,木村資生により提唱された30).発表された当時,中立進化説は古典的な自然選択説の支持者による激しい抵抗にあったが,現在では,生物種間の遺伝的な多様性はおもに中立進化により生じるということは広く受け入れられている.BEPモデルによるシミュレーションにてこのような中立進化が起こる条件においては,ひとつの細胞の変異プロファイルより多領域シークエンシングの解像度ではとらえきれない無数の中立変異を蓄積する微小なクローンが存在するであろうことも予測された.また,高い変異率の代わりにモデルを拡張してがん幹細胞の存在を仮定しても,同様に中立進化の起こりうることが見い出された.
最近,ほかの研究グループからも,筆者らと同様な結果が報告されている.大腸がんにおける腫瘍内不均一性を多領域シークエンシングと高深度シークエンシングを組み合わせて解析することにより,腫瘍の全体にわたる均一な腫瘍内不均一性が見い出された31).さらに,腫瘍のある部位に検出されたプログレッサー変異がそこからはなれた部位にアレル頻度の低いサブクローナルな変異として混ざり込む現象に注目しシミュレーションを用いて検証することにより,ビッグバンモデルが提唱された.このビッグバンモデルでは,がんの進化の初期に多くのサブクローンが生じ,そののち,それらが一部混ざり合いながら自然選択をうけずに広がり,腫瘍のどの部分においても一様に高い不均一性が形成されるとする.また,肝細胞がんにおける多領域シークエンシングのデータから得られたクローンの多様性に関する統計量の計測値が,解析的に導かれた中立進化が成り立つときに得られる理論値と統計的に一致することが示され,腫瘍内不均一性は中立進化により生じると主張された32,33).中立進化により無数の変異がひとつの腫瘍に蓄積されているとも主張され,これは筆者らの主張とも一致した.
3.これからの展開
ここまで,腫瘍内不均一性を理解するためのがんのゲノム解析および数理的な研究につき,最近の研究について解説した.ゲノム解析により種々のがんにおける広汎な腫瘍内不均一性の存在が示され,数理的な研究からは中立進化により腫瘍内不均一性が生じる可能性が示された.ここから,今後,期待される展開について述べる.
このレビューにおいては,ゲノム,とくに1塩基変異にしぼり腫瘍内不均一性について述べてきたが,オミックスのほかの階層においても同様な腫瘍内不均一性のあるものと思われる.筆者らも,大腸がんにおいて1塩基変異にくわえ,DNAコピー数,DNAメチル化,トランスクリプトームの腫瘍内不均一性が互いに相関しあいながら存在することを確認している19).ほかの研究グループからも,前立腺がんにおけるDNAメチル化の腫瘍内不均一性について報告されている34).また,最近,開発の進んでいる1細胞シークエンシングの技術を利用すれば,オミックスのおのおのの階層における腫瘍内不均一性を究極の解像度でみることが可能になるだろう35).たとえば,乳がんにおいてゲノムの1細胞シークエンシングが報告されている36).ゲノムの1細胞シークエンシングはDNAコピー数に関しては信頼性のあるデータが得られるようになっているものの,1塩基変異に関しては技術的なノイズの問題があり完全な1細胞の変異プロファイルを得るのはまだむずかしいのが現状である.そこで,複数の細胞において同定された1塩基変異の高深度シークエンシングにより,多くの変異が低い頻度で存在することが確認された.トランスクリプトームに関しては腫瘍からの試料の調製のむずかしさという問題はあるものの,1細胞RNA-seq法それ自体は技術的にほぼ完成されている.たとえば,グリオブラストーマにおいてトランスクリプトームの広汎な腫瘍内不均一性が1細胞RNA-seq法により明らかにされている37).DNAメチル化に関しても1細胞バイサルファイトシークエンシング法の開発が進んでおり,今後のがんの研究への応用が期待される38).
このような新しい実験技術の登場により,今後とも,腫瘍内不均一性に関するデータは量質ともにますます増えていくだろう.データが増加するにつれて,膨大なデータのなかの本質をとらえる数理的な研究の重要性が増してくる.これまでに得られているデータのなかからでも解かれるべき問題がある.たとえば,筆者らにより,大腸がんにおいて平行進化は観察されず腫瘍内不均一性は中立進化により説明される可能性のあることが示された一方で,腎臓がんおよびほかのがん腫においては平行進化がみられ,がんによっては腫瘍内不均一性の一部は自然選択により生じている可能性もある.筆者らは,BEPモデルによるシミュレーションにおいて,中立進化による無数のサブクローンをもつ腫瘍にくわえ,変異率が低く強いドライバー遺伝子のあるときには自然選択により均一な腫瘍が,変異率が低く弱いドライバー遺伝子が複数あるときには自然選択により複数のサブクローンをもつ腫瘍が生じることを見い出した.つまり,H2Oが温度や気圧というパラメーターの違いより氷,水,水蒸気の異なる相をとるように,がんもドライバー遺伝子の数,強さ,変異率などのパラメーターの違いにより異なる進化ダイナミクスの相を示すと考えられた.これらのパラメーターががんにより異なるのはもっともだと思われるし,同じがんの進化の過程においても変異率などは変化していくと想像される.今後,BEPモデルあるいはより抽象化した数理モデルを解析的に分析したり,種々のがんについてパラメーターのデータを収集したりすることにより,腫瘍内不均一性の生じる原理の詳細をさらにつめていく必要がある.
近年の計算資源の拡大とともに,とくにシミュレーションを中心とする計算科学の可能性が拡大している.筆者らの研究も,スーパーコンピューターを利用することによりはじめて可能となった.BEPモデルによるシミュレーションは確率的であり結果が試行ごとに異なるため,同じ条件の複数の試行の平均をみる必要があり,さらに,十分なパラメーター空間を探索するとなると膨大な計算時間を必要とするため通常の計算資源ではむずかしい.また.生物システムのシミュレーションのむずかしさのひとつとして,多くの内部パラメーターの値が実験的に直接には測定できず正確には決まっていないことがあげられる.そのような場合,観測データからパラメーター値を推定する必要があるが,そのようなパラメーターの推定の方法として,近年,実データとシミュレーションモデルがあたえられたときに実データを生じるパラメーター値の確率分布を計算できるapproximate Bayesian computation(ABC)法が注目されている39).もっとも簡単なかたちのABC法では,おのおののパラメーター値(複数のパラメーターがある場合にはその組合せ)について複数回のシミュレーションデータを生成し,データの特徴を記述する要約統計量を用いてシミュレーションデータと実データの類似性を評価して,おのおののパラメーター値において類似したシミュレーションデータが得られる確率を計算する.ベイズの定理より原因と結果を逆転させることができ,この確率は実データがあたえられたときにそのようなデータを生じることのできるパラメーター値の確率分布になる.筆者らも,BEPモデルによるシミュレーションにおいて,実データと同様の変異プロファイルを生じるパラメーター値の探索にABC法を用いた19).さらに,ABC法はパラメーターの推定だけでなくモデルの選択にも適用が可能である.つまり,ABC法を用いてあたえられたデータをよりよく再現できるようモデルを改良することができる.筆者らによるBEPモデルはまだトイモデルの域をでていないが,今後,ますます増えていくと思われる腫瘍内不均一性に関する実験データをABC法のようなデータ同化手法を用いて数理モデルに取り込んでいくことにより,より現実的ながんの進化のシミュレーションが行えるであろう.
がんの研究分野においては,20世紀後半の分子生物学の進展により発がんの分子機構が明らかにされ,現在,その知見をもとにさまざまな分子標的薬が開発されている.また,次世代シークエンサーを利用しさまざまながんにおいて変異のカタログ化が進行しており,今後,臨床における塩基配列の解読と分子標的薬とを組み合わせた個別化医療が進んでいくと考えられる.しかしながら,多くのがんにおいて治療抵抗性の問題は解決されておらず,治療により一時的に縮小した腫瘍も,多くの場合,結局は再発してしまう.筆者らによるシミュレーションの結果より,ひとつの腫瘍のなかに中立進化により生じた無数のクローンが存在していることが予測されたが,この結果は,がんの治療抵抗性の根本的な原因である可能性がある.中立な変異であるかどうかは環境にも依存するので,治療のまえには中立な変異であっても,治療により環境が変わればドライバー遺伝子の変異,つまり,治療抵抗性の変異になりうる.すなわち,どのような治療をしても無数のクローンのなかにそのような変異を獲得した治療抵抗性のクローンが存在してしまい,結局は再発にいたることが考えられる(図4).最近の大腸がんにおける抗EGFR抗体医薬の治療耐性の獲得に関する研究も,血中マーカーのダイナミクスの数理解析により,治療の開始の際にすでに治療抵抗性のクローンは存在しているという見方が支持されている40).
近年,治療抵抗性および感受性クローンを仮定したモデルを用いた数理的な研究により,抗がん剤の投薬の計画を調整することにより,がんの再発を遅延あるいは阻止させることのできる可能性が示されている41,42)(図5).たとえば,ひとつの腫瘍に多数の治療感受性のクローンと少数の治療抵抗性のクローンが存在する場合,通常のように大用量の抗がん剤を継続的に投与すれば治療感受性のクローンが減少することにより腫瘍は一時的に縮小するが,やがて,治療抵抗性クローンが増殖することによりがんは再発にいたる.一方で,もし用量を調整して治療感受性クローンをすべて殺さずに治療抵抗性クローンとうまく競合させることができれば,全体としてがんの成長を遅らせて患者の予後を伸ばすことが可能と思われる.また,HIVの治療において行われているように,複数の薬剤を使うことにより治療抵抗性のクローンの出現を抑えられることも示されている43).がんの治療において治療抵抗性の克服は最重要課題のひとつであるが,このような試みに腫瘍内不均一性の解析の研究から見い出された中立進化のモデルを組み込むことにより,解決の糸口がみえてくる可能性がある.
おわりに
これまで,がんの研究分野は分子生物学など個別的あるいは定性的な実験科学が主流であり,一部の例外をのぞき,理論科学および計算科学は華やかな実験科学の陰にかくれていた感がある.しかしながら,現在,次世代シークエンサーに代表される新技術の登場により包括的かつ定量的なデータの取得が可能になり,ようやく,理論科学および計算科学が表舞台に浮上し実験科学と融合する下地が整いつつある.がんの治療は日々進歩しているものの,がんはいまだ不治の病であることに変わりはない.筆者らは,がんを根本的に克服するためには異分野との融合研究によるがんの進化のシステムレベルでの原理的な理解が必須であると考えている.今後,さまざまな分野から研究者が参入し,がんの研究における異分野との融合が進むことを期待しつつ筆をおく.
文 献
- Nowell, P. C.: The clonal evolution of tumor cell populations. Science, 194, 23-28 (1976)[PubMed]
- Fearon, E. R. & Vogelstein, B.: A genetic model for colorectal tumorigenesis. Cell, 61, 759-767 (1990)[PubMed]
- Vogelstein, B., Papadopoulos, N., Velculescu, V. E. et al.: Cancer genome landscapes. Science, 339, 1546-1558 (2013)[PubMed]
- Garraway, L. A. & Lander, E. S.: Lessons from the cancer genome. Cell, 153, 17-37 (2013)[PubMed]
- Gerlinger, M., Rowan, A. J., Horswell, S. et al.: Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med., 366, 883-892 (2012)[PubMed]
- Gerlinger, M., Horswell, S., Larkin, J. et al.: Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet., 46, 225-233 (2014)[PubMed]
- Nik-Zainal, S., Van Loo, P., Wedge, D. C. et al.: The life history of 21 breast cancers. Cell, 149, 994-1007 (2012)[PubMed]
- Miller, C. A., White, B. S., Dees, N. D. et al.: SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput. Biol., 10, e1003665 (2014)[PubMed]
- Roth, A., Khattra, J., Yap, D. et al.: PyClone: statistical inference of clonal population structure in cancer. Nat. Methods, 11, 396-398 (2014)[PubMed]
- Hajirasouliha, I., Mahmoody, A., Raphael, B. J. et al.: A combinatorial approach for analyzing intra-tumor heterogeneity from high-throughput sequencing data. Bioinformatics, 30, i78-i86 (2014)[PubMed]
- Fischer, A., Vazquez-Garcia, I., Illingworth, C. J. et al.: High-definition reconstruction of clonal composition in cancer. Cell Rep., 7, 1740-1752 (2014)[PubMed]
- Zhang, J., Fujimoto, J., Zhang, J. et al.: Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science, 346, 256-259 (2014)[PubMed]
- de Bruin, E. C., McGranahan, N., Mitter, R. et al.: Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science, 346, 251-256 (2014)[PubMed]
- Yates, L. R., Gerstung, M., Knappskog, S. et al.: Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med., 21, 751-759 (2015)[PubMed]
- Cooper, C. S., Eeles, R., Wedge, D. C. et al.: Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat. Genet., 47, 367-372 (2015)[PubMed]
- Boutros, P. C., Fraser, M., Harding, N. J. et al.: Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet., 47, 736-745 (2015)[PubMed]
- Bashashati, A., Ha, G., Tone, A. et al.: Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J. Pathol., 231, 21-34 (2013)[PubMed]
- Suzuki, H., Aoki, K., Chiba, K. et al.: Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet., 47, 458-468 (2015)[PubMed] [新着論文レビュー]
- Uchi, R., Takahashi, Y., Niida, A. et al.: Integrated multiregional analysis proposing a new model of colorectal cancer evolution. PLoS Genet., DOI: 10.1371/journal.pgen.1005778[PubMed]
- Knudson, A. G. Jr.: Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA, 68, 820-823 (1971)[PubMed]
- Beerenwinkel, N., Antal, T., Dingli, D. et al.: Genetic progression and the waiting time to cancer. PLoS Comput. Biol., 3, e225 (2007)[PubMed]
- Haeno, H., Iwasa, Y., Michor, F. et al.: The evolution of two mutations during clonal expansion. Genetics, 177, 2209-2221 (2007)[PubMed]
- Bozic, I., Antal, T., Ohtsuki, H. et al.: Accumulation of driver and passenger mutations during tumor progression. Proc. Natl .Acad. Sci. USA, 107, 18545-18550 (2010)[PubMed]
- Iwasa, Y. & Michor, F.: Evolutionary dynamics of intratumor heterogeneity. PLoS One, 6, e17866 (2011)[PubMed]
- Durrett, R., Foo, J., Leder, K. et al.: Intratumor heterogeneity in evolutionary models of tumor progression. Genetics, 188, 461-477 (2011)[PubMed]
- Gonzalez-Garcia, I., Sole, R. V., Costa, J. et al.: Metapopulation dynamics and spatial heterogeneity in cancer. Proc. Natl. Acad. Sci. U A, 99, 13085-13089 (2002)[PubMed]
- Sottoriva, A., Verhoeff, J. J., Borovski, T. et al.: Cancer stem cell tumor model reveals invasive morphology and increased phenotypical heterogeneity. Cancer Res., 70, 46-56 (2010)[PubMed]
- Waclaw, B., Bozic, I., Pittman, M. E. et al.: A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature, 525, 261-264 (2015)[PubMed]
- Niida, A., Ito, S., Tremmel, G. et al.: Cancer evolution simulation identifies possible principles underlying intratumor heterogeneity. bioRxiv, 022806 (2015)
- Kimura, M.: Evolutionary rate at the molecular level. Nature, 217, 624-626 (1968)[PubMed]
- Sottoriva, A., Kang, H., Ma, Z. et al.: A Big Bang model of human colorectal tumor growth. Nat. Genet., 47, 209-216 (2015)[PubMed]
- Ling, S., Hu, Z., Yang, Z. et al.: Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc. Natl. Acad. Sci. USA, 112, E6496-E6505 (2015)[PubMed]
- Durrett, R.: Population genetics of neutral mutations in exponentially growing cancer cell populations. Ann. Appl. Probab., 23, 230-250 (2013)[PubMed]
- Brocks, D., Assenov, Y., Minner, S. et al.: Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep., 8, 798-806 (2014)[PubMed]
- Navin, N. E.: The first five years of single-cell cancer genomics and beyond. Genome Res., 25, 1499-1507 (2015)[PubMed]
- Wang, Y., Waters, J., Leung, M. L. et al.: Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature, 512, 155-160 (2014)[PubMed]
- Patel, A. P., Tirosh, I., Trombetta, J. J. et al.: Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 344, 1396-1401 (2014)[PubMed]
- Smallwood, S. A., Lee, H. J., Angermueller, C. et al.: Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods, 11, 817-820 (2014)[PubMed]
- Csillery, K., Blum, M. G., Gaggiotti, O. E. et al.: Approximate Bayesian Computation (ABC) in practice. Trends Ecol. Evol., 25, 410-418 (2010)[PubMed]
- Diaz, L. A. Jr., Williams, R. T., Wu, J. et al.: The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature, 486, 537-540 (2012)[PubMed]
- Gatenby, R. A., Silva, A. S., Gillies, R. J. et al.: Adaptive therapy. Cancer Res., 69, 4894-4903 (2009)[PubMed]
- Ideta, A. M., Tanaka, G., Takeuchi, T. et al.: A mathematical model of intermittent androgen suppression for prostate cancer. J. Nonlinear Sci., 18, 593-614 (2008)
- Bozic, I., Reiter, J. G., Allen, B. et al.: Evolutionary dynamics of cancer in response to targeted combination therapy. Elife, 2, e00747 (2013)[PubMed]
著者プロフィール
略歴:2007年 東京大学大学院理学系研究科 修了,同年 東京大学分子細胞生物学研究所 研究員,2008年 東京大学医科学研究所 研究員を経て,2011年より同 特任助教(現 助教).
研究テーマ:がんのバイオインフォマティクスおよび数理解析.
抱負:実験と数理,および,基礎と臨床の共同研究をつうじて,がんの進化の原理の解明,治療抵抗性の克服,がんの進化のシミュレーションの臨床応用をめざしている.
三森 功士(Koshi Mimori)
九州大学病院別府病院 教授.
宮野 悟(Satoru Miyano)
東京大学医科学研究所 教授.
© 2016 新井田厚司・三森功士・宮野 悟 Licensed under CC 表示 2.1 日本
