精密な定量プロテオミクスにもとづく生命科学の研究
2017/04/11
松本雅記・中山敬一
(九州大学生体防御医学研究所 プロテオミクス分野)
email:松本雅記,中山敬一
領域融合レビュー, 6, e002 (2017) DOI: 10.7875/leading.author.6.e002
Masaki Matsumoto & Keiichi I. Nakayama: Life science relying on accurate and reproducible quantitative proteomics.

近年の質量分析計のいちじるしい高性能化および種々の定量技術の開発によりプロテオーム解析の技術は成熟し,いちどに数千のタンパク質の同定および定量が可能になり生命科学のさまざまな領域の発展に大いに貢献している.その一方で,現在,普及している手法は定量性や再現性の問題に直面しており新たな方法論の構築が望まれてきた.近年,従来のプロテオーム解析法の問題点を解消できる手法として多重反応モニタリング法などの技術を用いたターゲットプロテオミクスが確立され,任意のタンパク質のハイスループットかつ正確な定量が可能になった.しかしながら,多重反応モニタリング法を実施するために必要な情報リソースの不足およびその測定メソッドの構築の煩雑さから,広く普及するにはいたっていない.最近,筆者らを含むいくつかの研究グループから,大規模なターゲットプロテオミクスを容易に可能にする技術開発が報告され,ようやく精密な定量プロテオミクスにもとづく生命科学の研究がスタートしつつある.このレビューにおいては,質量分析計を用いたプロテオーム解析の原理および特徴を説明するとともに,最新のターゲットプロテオミクスの動向を筆者らによる最近の取り組みもまじえて解説し,さらに,がん代謝の研究への応用について紹介する.
ヒトなど主要な生物種のゲノム情報の解読を背景として,さまざまなオミクス解析の技術が発展し,網羅的な分子計測データから生命現象をシステムとして理解する試みがなされている.とくに,次世代シークエンス技術の登場はハイスループットかつ網羅性の高い塩基配列の情報の取得を可能にし,さまざまな生命現象や疾患とゲノム,エピゲノム,トランスクリプトームとの関係性が明らかにされている.その一方で,これら遺伝情報と生命現象とをつなぐ原理や法則の蓄積は乏しく,いまだ生命システムへの本質的な理解からはほど遠い状況にある.たとえば,がんはさまざまなゲノムの変異を原因として生じるが,これらの変異がいかにしてがん細胞のさまざまな特徴を生み出すのかについてはほとんどわかっていない.すなわち,生命システムの表現型,たとえば,がんの増殖の速度や運動能などと,さまざまな核酸分子のプロファイルとのあいだには巨大なブラックボックスが存在する.このようなブラックボックスの解消のためには,表現型と相関する分子シグネチャーの同定が必須である.なかでも,生命現象の直接的な担い手であるタンパク質の量的あるいは質的な変化をグローバルにとらえるプロテオーム解析の重要性は明白であり,ゲノム情報の整備および質量分析計の高性能化を追い風に大きな期待がよせられてきた1).
現在,液体クロマトグラフィー(LC)とタンデム質量分析計(MS/MS)とを連結した,いわゆるLC-MS/MSによるプロテオーム解析法がもっとも普及している.LC-MS/MSによるプロテオミクスは,その原理からノンターゲットプロテオミクスとターゲットプロテオミクスの2つに大別される2)(図1).また,タンパク質を酵素により消化して得られるペプチドを解析するボトムアッププロテオミクスと,タンパク質を消化せずにそのまま解析するトップダウンプロテオミクスとに分類することもできるが(図1),このレビューでは,現時点においてより実用的なボトムアッププロテオミクスについてのみ記述する.なお,ボトムアッププロテオミクスは,より慣用的かつ普及しているショットガンプロテオミクスとよぶことにする.
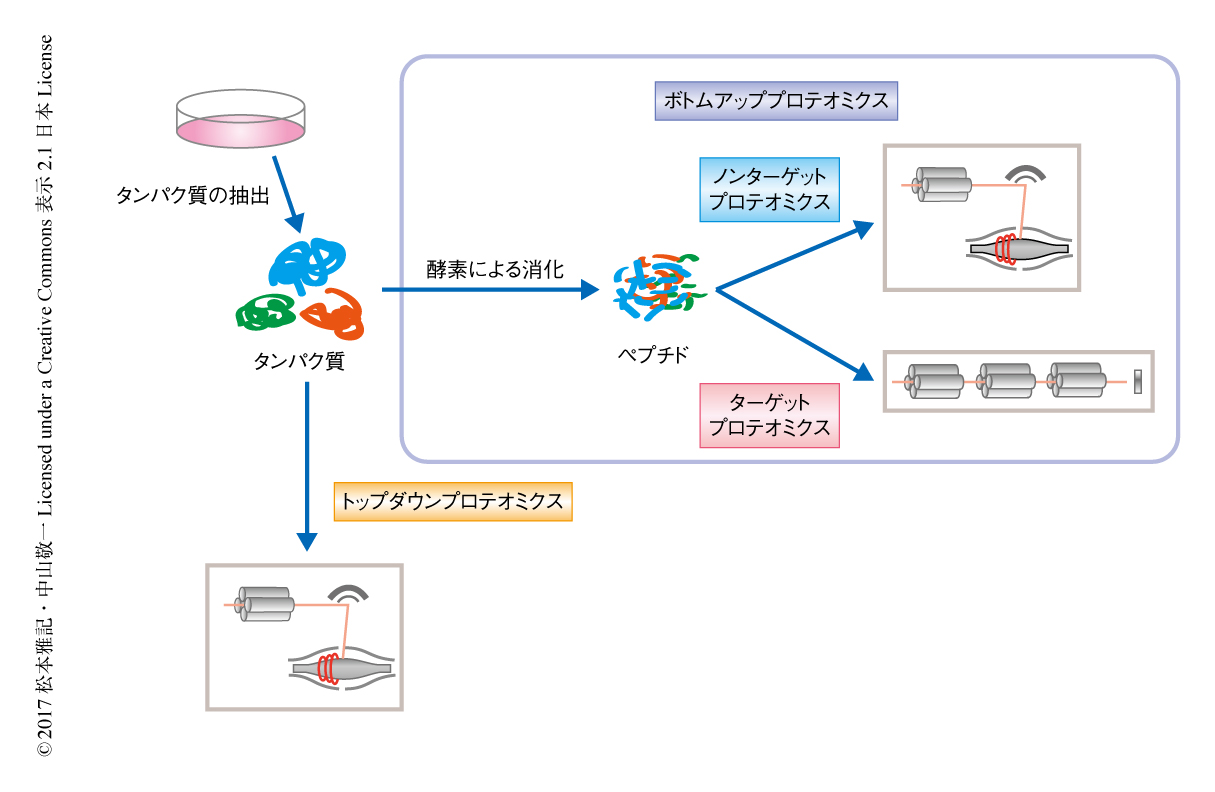
このレビューにおいては,ノンターゲットプロテオミクスおよびターゲットプロテオミクスの原理およびその特徴について解説するとともに,近年,急速に進んだターゲットプロテオミクスの大規模化の動向,さらに,がん代謝の研究への応用について紹介する.
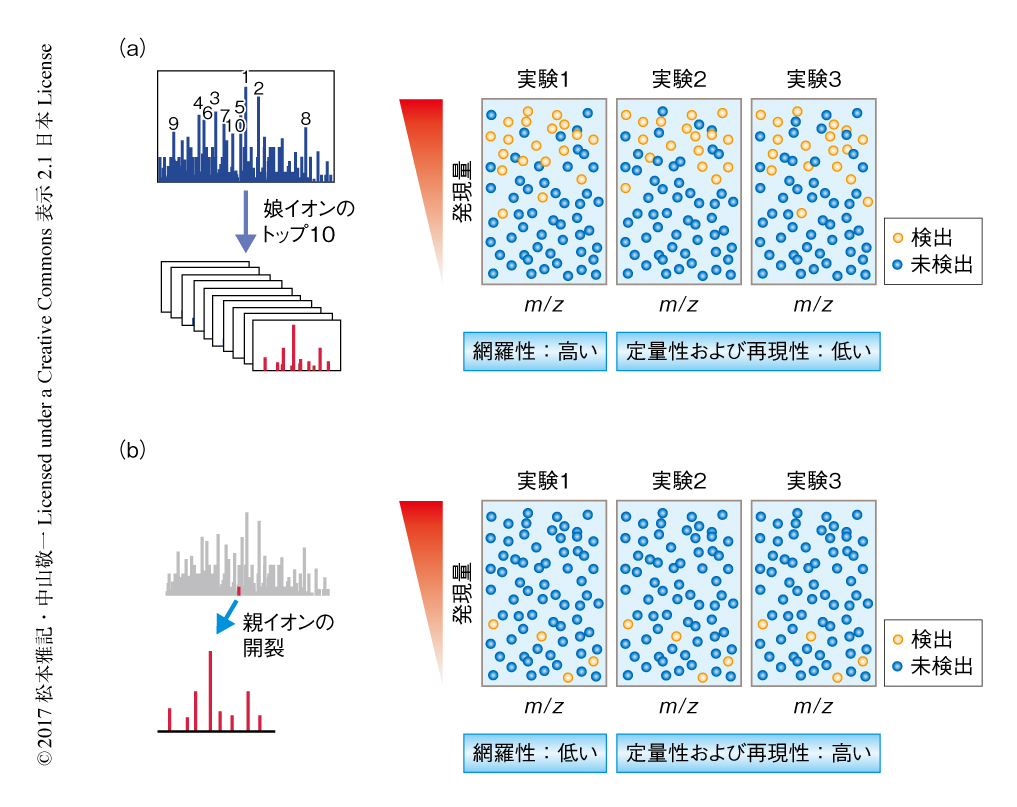
現在,ショットガンプロテオミクスのなかでもっとも普及しているのは,データ依存的解析(data-dependent acquisition:DDA)法によるノンターゲットプロテオミクスである.通常,複雑なペプチドの混合物は液体クロマトグラフィーにより分離してもなお多数のペプチドが同時に質量分析計に導入されるため,ひとつのMSスペクトルから多数のペプチドに由来するシグナルが検出される.データ依存的解析法においては,MSスペクトルから検出される多数のシグナルのうち強度の強いものを親イオンとして自動的に選択し,衝突誘起解離(collision-induced dissociation:CID)法によりMS/MSスペクトルを取得する(図2).得られたMS/MSスペクトルはペプチドの配列を反映する断片イオンの質量の情報を含む.通常,タンパク質はトリプシンなどのアミノ酸配列に特異的な酵素により消化されるため,親イオンの質量をもとに,タンパク質の配列のデータから候補となるペプチドの配列が絞り込まれる.候補となるペプチドの配列から理論的なMS/MSスペクトルが得られるが,これを実測されたMS/MSスペクトル(ペプチド結合がランダムに切断された場合に生じる断片イオン)と照合し,パターンの一致するペプチドの配列を同定する.ノンターゲットプロテオミクスは興味のある検体に含まれるタンパク質の同定を目的としており,いちどに多数のタンパク質を検出できるため,おもに探索的な研究に利用されている.近年では,ノンターゲットプロテオミクスに定量法を組み合わせることにより,より定量的な解析が可能になっている.定量の技術としては,スペクトルカウント法,標識フリー定量法,in vivo標識法,in vitro標識法があげられる.
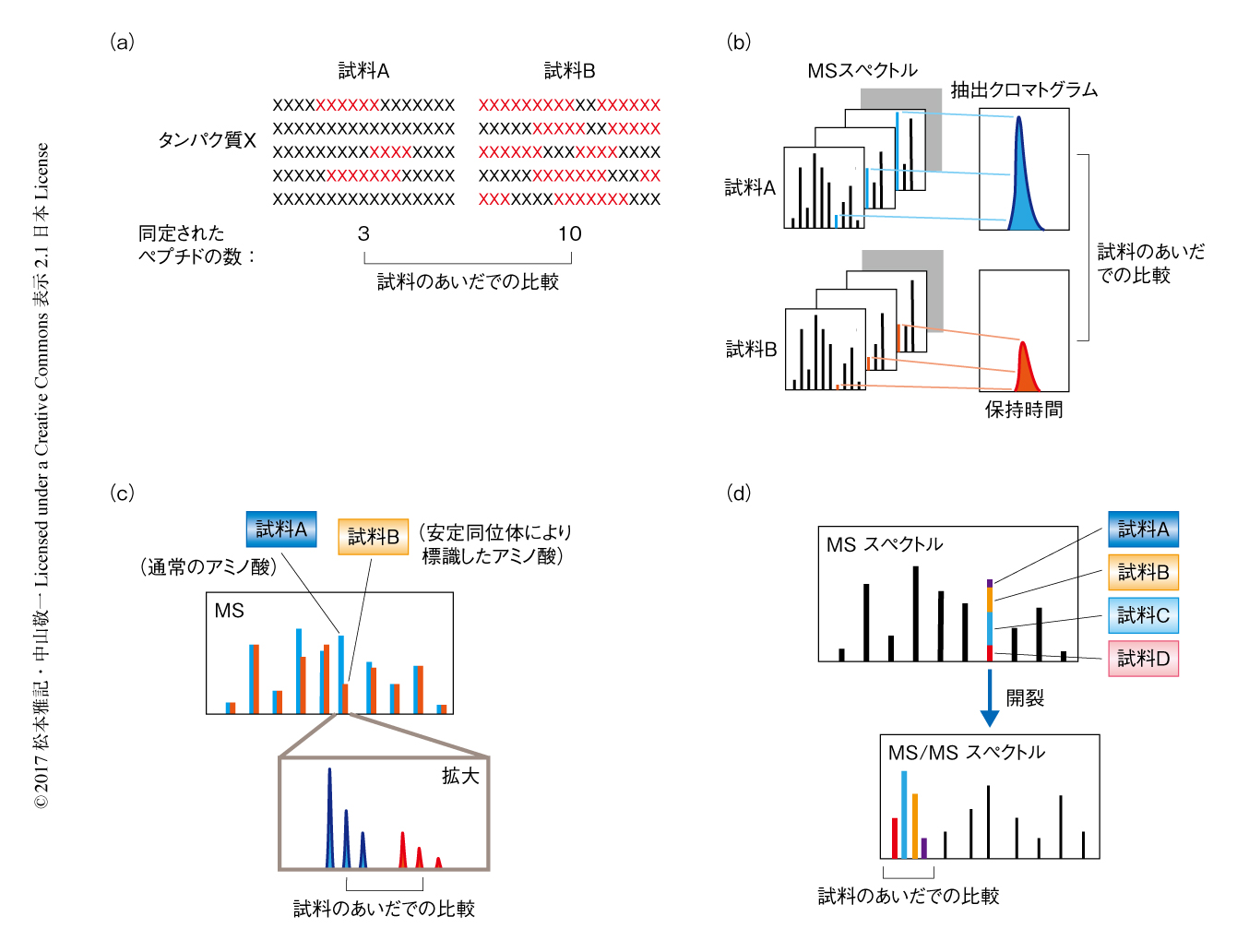
スペクトルカウント法(図3a):ノンターゲットプロテオミクスにおいては,おのおののタンパク質において同定されるペプチドの数はタンパク質の存在量におおむね依存する.つまり,同定されたペプチドの数を比較することにより,おのおのの検体のあいだの発現量の比較が可能である.スペクトルカウント法は使用する装置の分解能や質量精度に依存しないため,もっとも汎用性の高い手法である.また,同定されたペプチドの数をタンパク質の分子量や理論ペプチド数で標準化する手法や,これを拡張したexponentially modified protein abundance index(emPAI)などが開発され3),比較定量のみならずタンパク質の存在量の推定にも利用されている.
標識フリー定量法(図3b):個々のペプチドイオンのシグナルからクロマトグラムを得ることによりペプチドの存在量を概算する手法である.特別な前処理が不要であり,あらゆる試料に適用できる手軽な手法である.ただし,特定のペプチドの抽出クロマトグラムを正確に得るためには高分解能かつ高質量精度の質量分析計を必要とする.また,定量性を保証するためには基本的にシングルショット(前分画を施さないLC-MS)での解析が前提であり,比較的発現量の高いタンパク質が解析の対象となる.
in vivo標識法(図3c):培養細胞などを培養する際に安定同位体により標識した物質を取り込ませることで代謝的にタンパク質を標識する.とくに,安定同位体により標識したアミノ酸を用いる方法が普及しており,SILAC(stable isotope labeling with amino acid in cell culture)法とよばれている4).細胞の発現するすべてのタンパク質が安定同位体により標識されるため,試料の調製の初期段階において検体を混合することができ,試料の調製の過程におけるブレを最小限に抑えることが可能である.したがって,オルガネラの画分やアフィニティー精製など複雑な試料の調製を必要とする実験法との相性がよい.
in vitro標識法(図3d):代謝的な標識が適用できない臨床検体などを標識するのに,安定同位体を含む試薬を用いてタンパク質あるいはペプチドを修飾する手法である.個々の検体を異なる質量をもつ試薬により標識するnon-isobaricタグ法5) と,異なる質量をもつリポーター部位とバランサー部位とを組み合わせて同一の質量をもたせたisobaric試薬により標識するisobaricタグ法6) とに分けられる.non-isobaricタグ法においてはおのおのの検体に由来するペプチドが異なる質量をもつ親イオンとして観測され,そのシグナルの強度から比較定量が可能である(質量分析計における定量の原理は,SILAC法と同じ).一方,isobaricタグ法においては標識ののちのペプチドの質量はまったく同一となり,MSスペクトルにおいては単一のピークとして検出される.このペプチドは衝突誘起解離法によりリポーターとバランサーとのあいだが切断されるため,MS/MSスペクトルの低質量の領域に質量が1 Daずつ異なるリポーターイオンが検出され,これらのシグナルの強度がそのペプチドの試料のあいだの相対量比を示す.
各種の定量の技術の普及と近年の質量分析計の急速な性能の向上により,ノンターゲットプロテオミクスにより大腸菌など比較的単純なプロテオームに含まれるほとんどのタンパク質の検出が可能になった7).さらに,ヒトやマウスなどより複雑なプロテオームについても,LC-MS/MS解析のまえに分画を施すなどのくふうにより数千から1万程度のタンパク質の同定が可能である8,9).ヒトの培養細胞においてmRNAとして発現の確認される遺伝子は通常10,000~12,000であることを考えると8),ほぼ網羅的なプロテオーム発現の解析が可能になっている.このような技術的な成熟を反映してか,近年,2つの研究グループより,ヒトの全プロテオームのドラフトが報告された10,11).いずれの論文も,公共リポジトリに登録された質量分析計のデータに,おのおのの研究グループにおいて新たに取得したデータをくわえ,統一的な解析パイプラインにより処理することで結果の統合を図っている.それぞれ,18,097種類および17,294種類のタンパク質を同定しており,ヒトのゲノムに存在するタンパク質をコードする遺伝子の約9割が実在のタンパク質として発現が確認されたことになる.さまざまな計測条件において取得されたデータが混在しているため,スペクトルカウント法や標識フリー定量法を用いてタンパク質の発現量を大まかに推定するにとどままっているが,ヒトのプロテオーム発現の全体像が得られた意義は大きい.その一方で,これらの報告は,データ依存的解析法による多数のデータを集積させたことで多くの偽陽性ヒットを含む可能性が指摘されており12),多数のタンパク質の存在がいまだに実験的に証明されていない可能性がある.このようなデータ依存的解析法による多数のデータを統合する際の新たな枠組みの開発などが今後の重要な課題であろう.たとえば,2015年より,わが国初の公共プロテオーム情報データベースであるjPOST(Japan Proteome Standard Repository/Database)の開発がはじまり(URL:http://jpostdb.org/),今後,より高質かつ信頼性の高いプロテオーム情報の公開が期待されている.
このように,データ依存的解析法によるノンターゲットプロテオミクスが進む一方で,技術的な制約による問題点もうきぼりになっている.データ依存的解析法により同定されるタンパク質の数は取得したMS/MSスペクトルの数に依存するため,網羅性をあげるには必然的に長い分析時間を要する.すなわち,網羅性とスループットには完全にトレードオフの関係がある.最近,普及しているフーリエ変換型ハイブリッド質量分析計を用いてデータ依存的解析法により2時間程度のデータの取得を実施すると約2000種類のタンパク質が同定されるが,このなかには細胞骨格タンパク質やリボソームタンパク質など発現量が高いものが多く,シグナル伝達タンパク質や転写因子などはまばらである.これは,データ依存的解析法によるMS/MSスペクトルの取得がタンパク質の存在量の多い順に実施されることに起因しており,発現量の低いタンパク質の検出をもとめる場合には1試料あたり4時間から6時間以上の分析時間を要する.つまり,データ依存的解析法では高スループットかつ発現量の低いタンパク質にアクセスが可能な解析を実施することは困難である.さらに,複雑な試料においてデータ依存的解析法を実施した場合,MS/MSスペクトルの取得がランダムになされるうえ,すべてのMS/MSスペクトルが高品質で得られるわけでないため,測定ごとに同定されるペプチドのリストが異なり,再現性の面においても大きな問題となる.
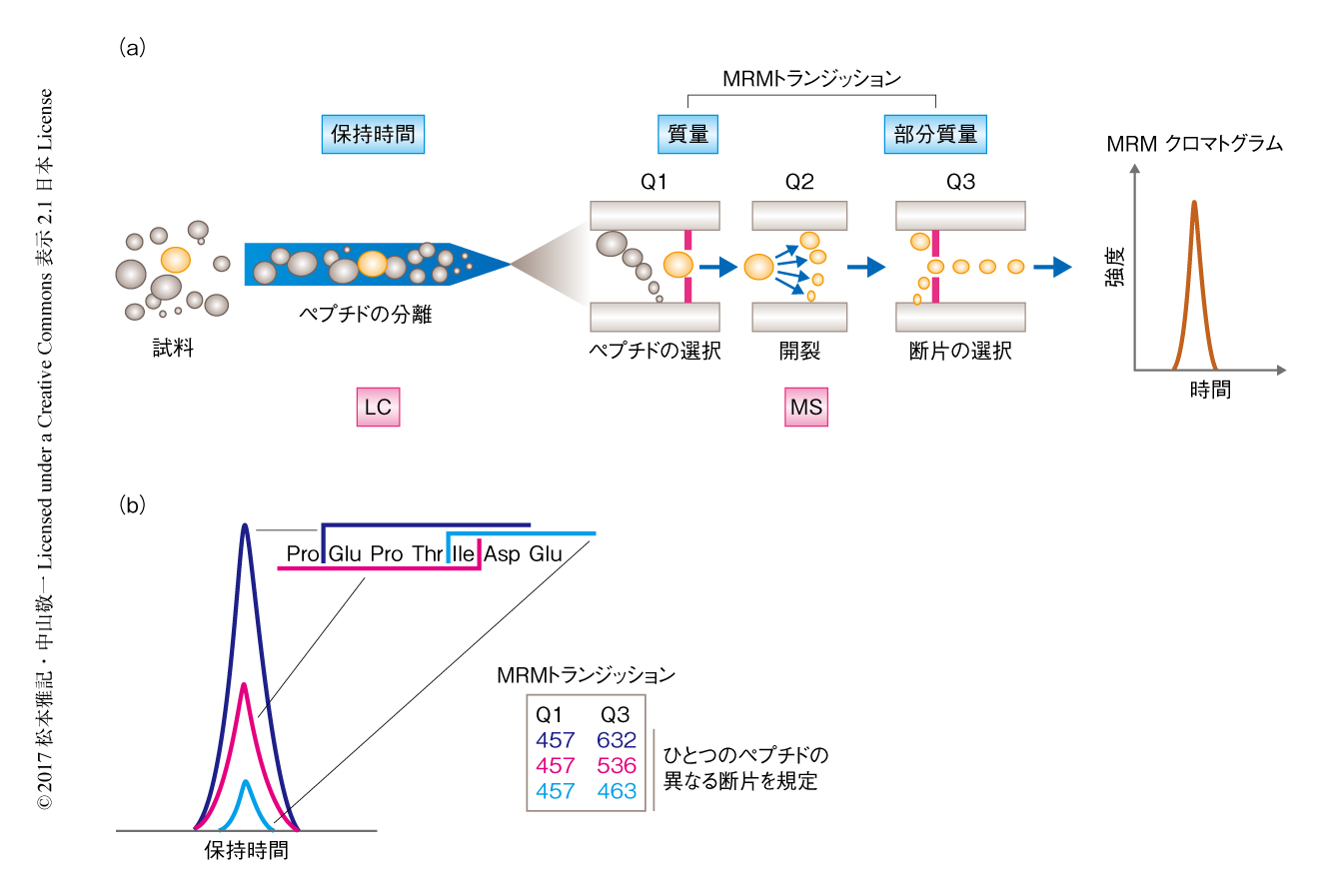
データ依存的解析法における問題や限界を相補する手法として,ターゲットプロテオミクスがきわめて有効である13-15).ターゲットプロテオミクスでは三連四重極型質量分析計を用いた多重反応モニタリング(multiple reaction monitoring:MRM)法(あるいは,選択反応モニタリング法,selected reaction monitoring:SRM法)を用いるのが一般的である.多重反応モニタリング法では,親イオンを通すQ1フィルターと開裂ののちの断片イオンを通すQ3フィルターの組合せ設定(MRMトランジッション)により,特定のイオンを特異的に検出かつ定量する(図4a).通常,ひとつのペプチド(Q1)に対して複数の断片(Q3)のMRMトランジッションを設定するが,これらはクロマトグラムにおいて完全に共溶出するピークとして検出される(図4b).実際の測定においては近傍にノイズのシグナルが生じることも多いが,これら共溶出を指標に目的とするペプチドのピークを同定できる.多重反応モニタリング法はいちどに計測できるペプチドの数に制限があるが,広いダイナミックレンジと高い選択性をもつことから,比較的発現量の低いタンパク質でも事前の分画なしに再現性よく定量することが可能である.また,安定同位体による標識を施した濃度が既知の内部標準を添加することにより正確な絶対定量も可能である.たとえば,出芽酵母において細胞あたり数十~106分子という広範な発現量のレンジに属する100種類のタンパク質を対象として絶対量の計測が実施されている16).このような高速性や定量性などの多重反応モニタリング法の利点は,ノンターゲットプロテオミクスにおいて見い出された興味あるタンパク質の評価の手段としてきわめて重要であり,実際,生体試料におけるバイオマーカーの探索の検証などに活用されている.
最近では,観測されるすべての(あるいは,一定の質量のレンジの)イオンをまとめてMS/MSスペクトルを取得するデータ非依存的解析(data-independent acquisition:DIA)法とよばれる方法が開発され普及しはじめている17).データ非依存的解析法により取得したデータのなかには,理論的にはすべての親イオンに由来する混合MS/MSスペクトルがクロマトグラムの溶出にしたがい格納されている.したがって,任意のペプチドに関するプロダクトイオンの抽出イオンクロマトグラム(すなわち,高分解能の擬似MRMクロマトグラム)を,事前に特定のペプチドに対する測定メソッドを組むことなく,データを取得したのちに表示することが可能である.しかしながら,データ非依存的解析法は多重反応モニタリング法による解析に比べ感度面で若干劣る状況であり,発現量の低いタンパク質の定量にはより高感度な装置の開発が望まれる.
近年では,質量分析計の高速化やスケジュール化(液体クロマトグラフィーにおける保持時間の指定)の発達により,より多成分に対する多重反応モニタリング法による解析や数百のペプチドの一斉定量が可能になっている.これまで,生命科学の研究において,免疫ブロット法などを用いてさまざまな条件におけるタンパク質の発現の挙動を計測することにより“タンパク質Xが生命現象Yにかかわる”という仮説が検証されてきたが,これを多成分で実施できる意義はきわめて大きい.多くのタンパク質はさまざまな基準により機能単位にカテゴライズされる.たとえば,解糖系やTCA回路などの代謝モジュールはほとんどが100種類以下のタンパク質から構成される.主要なシグナル伝達経路も同様におおむね100種類のタンパク質から構成される.また,タンパク質の機能単位でみても,多くの場合は数十~数百のタンパク質のメンバーから構成されるファミリーである.したがって,数十~数百のタンパク質を確実に一斉定量できれば,経路や機能単位の情報の取得が可能になる.
近年,多重反応モニタリング法を基盤としたターゲットプロテオミクスを大規模な解析に利用しようとする機運が高まり,そのためのインフラの構築が世界中で進められてきた18-20).多重反応モニタリング法においては,事前にタンパク質に特異的かつ高感度な対象ペプチド(proteotypic peptide:PTP)の選定,および,おのおののペプチドに対するMRMアッセイ(MRMトランジッションの組合せ)の作成および検証が必要である(図4b).PTPはおのおのの研究者が自身で同定したタンパク質のペプチドから選ぶことも可能であるが,現在では,データ依存的解析法により世界中で取得されたデータが各種のリポジトリサイトを介して集約され,高頻度に同定されるペプチドがその候補として利用されており,多重反応モニタリング法の普及を促進する重要な基盤となっている21).しかしながら,これらの情報はあくまでPTPの候補であり,実際には,これらのなかからより感度の高いペプチドの選定や測定に必要な情報(どの断片イオンを使うか,液体クロマトグラフィーにおける保持時間の情報など)を合成ペプチドなどの標品を用いて取得し,MRMメソッドの有効性を評価する必要があり,これが実際に多重反応モニタリング法を大規模に実施しようとした際に大きな障壁となっている.
MRMメソッドの構築の迅速化のため,ハイスループットなペプチド合成法により作製したペプチドライブラリーを用いた大規模なMRMメソッドの評価が実施されている.このような試みは,比較的ゲノムサイズの小さい出芽酵母や結核菌などから実施され,その戦略が確立されてきた22-24).最近,同様の手法によりヒトのプロテオームを対象とした網羅的なMRMメソッドが構築され,データベースが公開されている25)(human SRMAtlas resource,URL:http://www.srmatlas.org).このデータベースにおいては,実試料から同定されたペプチドにくわえ,バイオインフォマティクスによる予測により16万以上のPTPの候補の合成ペプチドを作製しMRMメソッドが精査されている.さらに最近では,大規模な合成ペプチドライブラリーの構築をめざしたプロジェクトProteomeTools(URL:http://www.proteometools.org)の一部(35万以上のペプチド)に関する報告がなされた26).ここでは,さまざまな機種あるいはイオンの開裂法によりMS/MSスペクトルを取得しているが,多重反応モニタリング法による評価は実施していない.その一方で,将来的に,トリプシン以外の酵素により生じるペプチドや修飾ペプチドまで拡張することを明言しており,将来的に,より網羅性の高いスペクトルライブラリーの充実がみこまれる.
このように,合成ペプチドを利用したMRMメソッドの構築が進められてきた一方で,合成ペプチドを用いるターゲットプロテオミクスに対する警鐘も鳴らされている27).おのおののタンパク質からの酵素消化によるペプチドの切り出しの効率が必ずしも完全ではない点や28),予測にもとづき合成された候補のペプチドが必ずしも高感度なペプチドである保証がないことなどから,MRMメソッドの構築に組換えタンパク質の利用が提案されてきた27,29).しかしながら,一般の研究室では組換えタンパク質の作製や精製は個々に実施されており,時間と労力を要するものであった.そのため,網羅的な組換えタンパク質を用いた大規模なMRMメソッドの構築やその評価は実施されていなかった.
筆者らは,Gatewayシステムに組み込まれたヒトの完全長cDNAライブラリーを用いた大規模な組換えタンパク質リソースであるin vitro proteomeに着目し30).質量分析に適した試料の調製法を確立した.最終的に,18,000種類以上のゲノムワイドな組換えタンパク質に対するMS/MSスペクトルの取得(200,000以上のペプチド)に成功した31).さらに,これらのMS/MSスペクトルの情報を利用して網羅的にMRMメソッドを構築し,実際の測定にもとづき精査することにより,ゲノムワイドなターゲットプロテオミクスを実施するための基盤情報の取得を完了した.さらに,組換えタンパク質を酵素消化した産物にはmTRAQ法による安定同位体の質量タグを付加しており,これらのリソースはMRMメソッドの構築のための情報源のみではなく,タンパク質の絶対定量の際の内部標準としても利用が可能である31).また,多重反応モニタリング法による計測に必要な情報を格納したデータベースと定量解析のためのツールも独自に作成しており,これらを含めたプラットフォームをiMPAQT(in vitro proteome-assisted MRM for protein absolute quantification)と名づけて公開している(URL:http://impaqt.jpost.org,図5).このデータベースでは,さまざまな分子機能,関与する生命現象,生化学的な経路にもとづいて任意のタンパク質を検索し,対応するMRMメソッドを取得することが可能である.
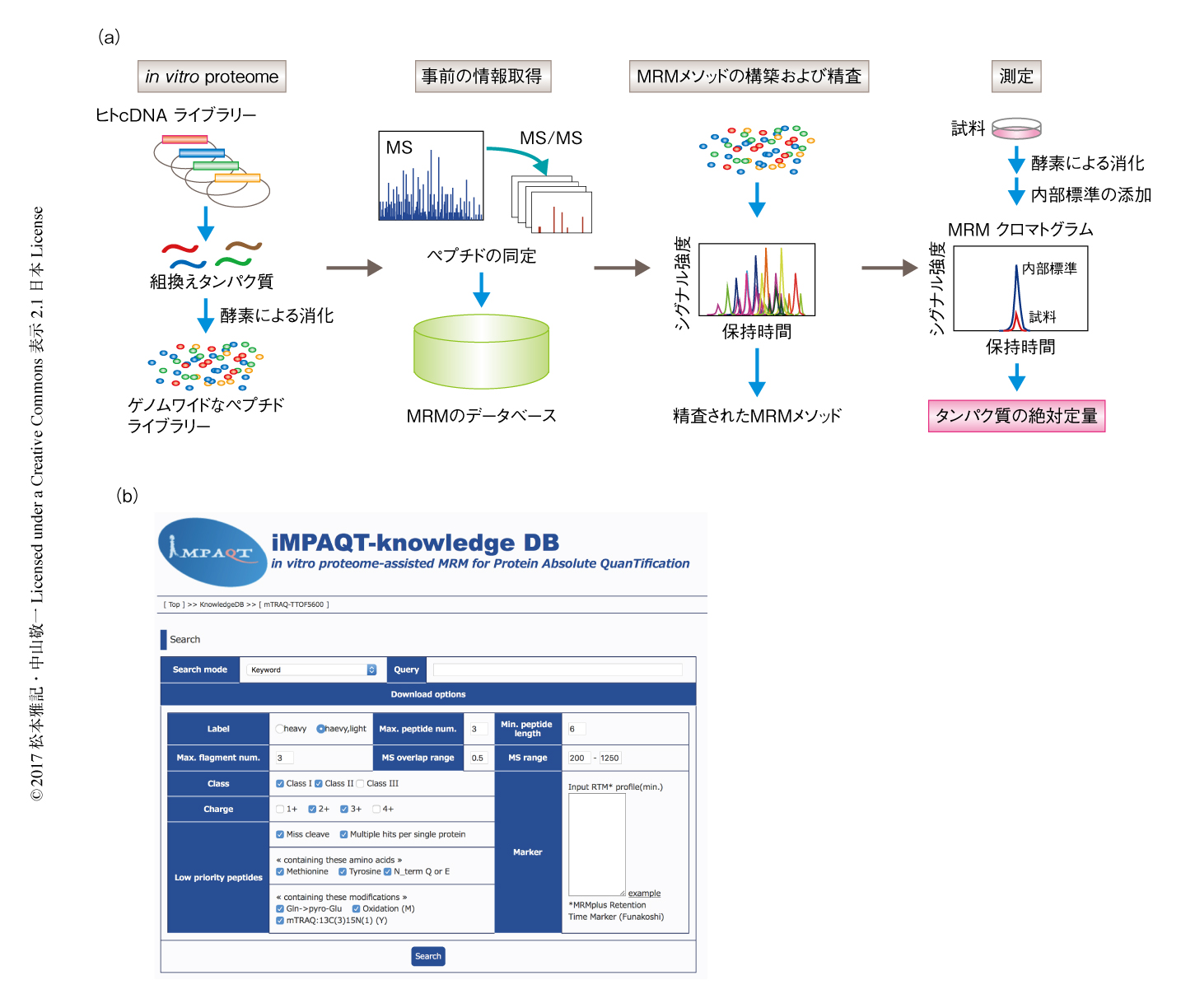
実際に,このデータベースを利用したところ,代謝経路に関連する約1000種類の酵素に関するMRMメソッドの設計および最適化は数日で完了し実試料の分析に進むことができた.そこで,すべての代謝酵素を対象とした定量代謝マップの作成に取り組み,ヒトの正常な線維芽細胞において発現する約650種類の代謝酵素の発現絶対量の計測に成功した31).その結果,解糖系の後半の反応にかかわるグリセルアルデヒド-3-リン酸デヒドロゲナーゼや乳酸デヒドロゲナーゼAなどはきわめて発現が高いのに対し,解糖系の律速段階であるヘキソキナーゼやホスホフルクトキナーゼは発現量の低いことが判明した.これらの酵素の発現量を低く抑えることが反応律速をつくりだすのに寄与する可能性が示唆された.このように,代謝経路の定量的な特徴は個々の酵素の機能や経路における位置を反映するのかもしれない.
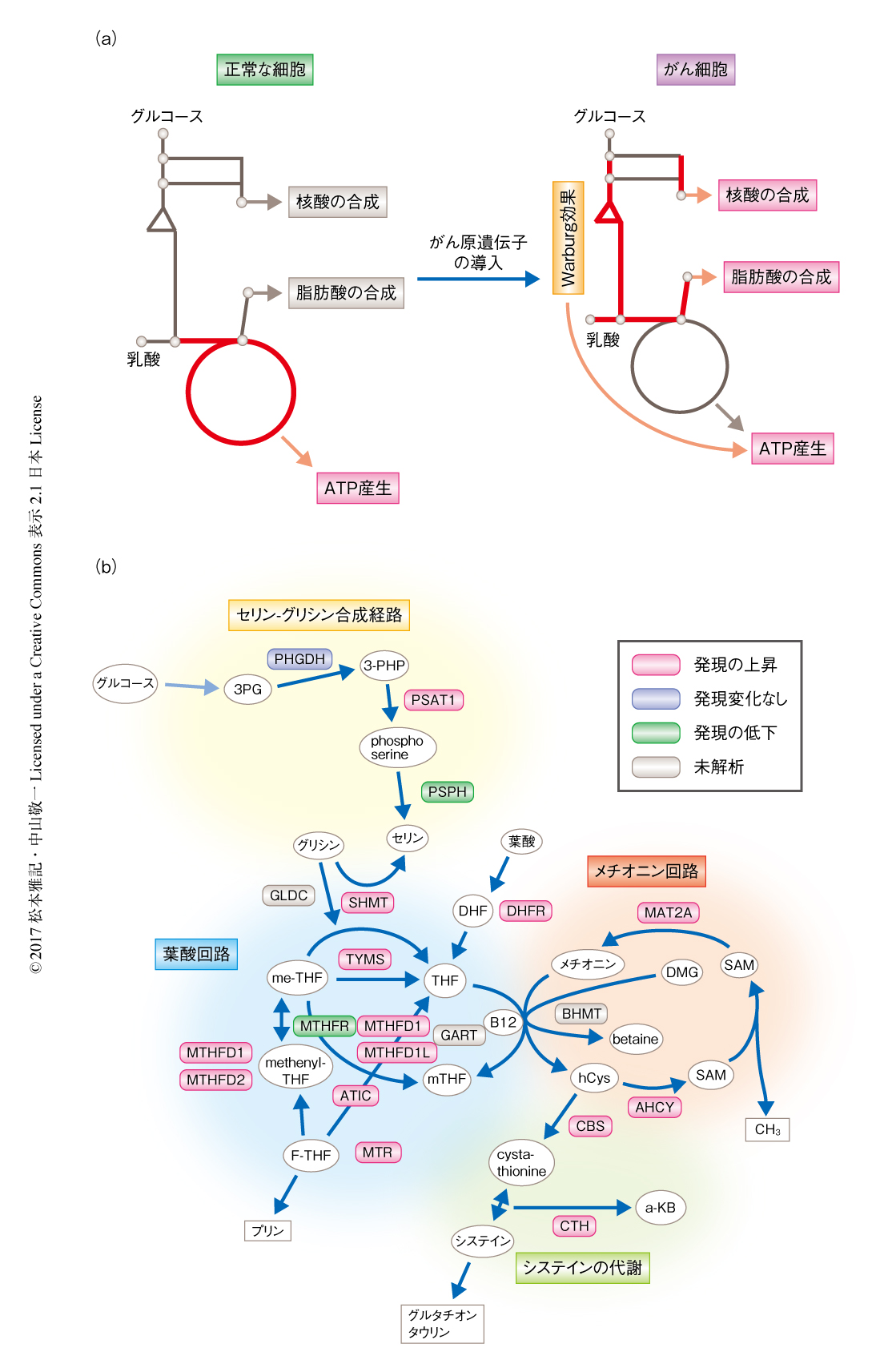
古くから,がんと代謝のあいだには密接な関係性が知られている.その代表的なものが,好気的な条件における解糖系の亢進,Warburg効果である32,33).このがんにおける解糖系の亢進は非代謝性のグルコースアナログを利用した陽電子断層撮像法(positron emission tomography:PET)としてがんの画像診断に応用されていることからも,多くのがんにおいて普遍的であることが広く認知されている34).しかしながら,どのような機構によりがんに特有の代謝状態が生じるのかについては不明な点も多い.近年,c-Myc遺伝子,HIF1遺伝子,p53遺伝子などがんに関連する遺伝子の変異や発現量の変化がさまざまな代謝酵素の発現に直接的に作用し,代謝経路を変化させていることが明らかにされ注目されている32,35,36).たとえば,c-Myc遺伝子は乳酸デヒドロゲナーゼA,ヘキソキナーゼ2,ピルビン酸キナーゼM2など解糖系に関連する多くの酵素の発現を上昇させる36).低酸素に応答性の転写因子HIF1の活性化は解糖系に関連するほとんどの酵素の発現を上昇させる.また,がん抑制遺伝子であるp53遺伝子は,グルコーストランスポーターやヘキソキナーゼ2など解糖系に関連する酵素の発現の抑制や,解糖系を負に制御するTIGARの発現の亢進により解糖系を抑制する37).さらに,p53はグルコース-6-リン酸脱水素酵素の発現の抑制を介したペントースリン酸回路の抑制やシトクロムcオキシダーゼ合成酵素2の発現の亢進による酸化的リン酸化の促進なども担う37).したがって,p53の代謝制御能の喪失は相対的に解糖系の活性化をひき起こすと考えられている.実際のがんにおいては,これらがんに関連する遺伝子の変化は複合的であり,その結果として生じる代謝経路の再編成はきわめて広範囲かつ複雑である可能性が高い.
代謝は多数の酵素の織りなす生化学反応の集積であり,細胞システムは内外の環境の摂動に応じて代謝経路をダイナミックに再編することによりさまざまな環境に適応する.がんに関連する遺伝子が代謝に直接的に影響をあたえることにくわえ,がん細胞は生体において微小環境の要因の影響も大いにうけると考えられるため,がん化にともなう代謝システムの変化はきわめて広範囲かつ多様な様相を示すと考えられている.代謝システムの状態を記述するには細胞における代謝酵素の活性を測定することがもっとも直接的であるが,現時点において,代謝酵素の活性を網羅的に計測する手立ては存在しないため,代謝酵素の発現量から酵素活性を推定するのが現実的である.したがって,がん代謝の研究への発現プロテオーム解析の適用の重要性は早くから認識されており,実際に,さまざまな研究が展開されてきた38).たとえば,初期のプロテオーム研究においては,2次元電気泳動を用いてさまざまながんの臨床検体を対象としたタンパク質の発現量の比較がなされており,がんにおいて発現の上昇するタンパク質のスポットとして解糖系に関連する多くの酵素が同定されている38-40).そののち,ショットガンプロテオミクスが普及し,がん細胞においてより広範な代謝酵素の発現量の変化が生じていることが示された41,42).近年では,大規模なプロテオームデータの取得とほかのオミクスデータとの統合により,がんにより生じるゲノムの変化とプロテオームとのあいだの関連に着目した研究もなされている43-45).
このようにがんのプロテオーム解析が世界中で進むなか,がん代謝の理解はどのくらい進んだのだろうか? 確かに,いちどに数千のタンパク質を同定し比較できるようにはなったが,さきに述べたように,この手法は再現性や定量性においていまだ問題点も多い.とくに,情報量が膨大かつノイズの多いタンパク質のリストのデータ解析においては,多くのデータを切り捨て特徴的な挙動を示すタンパク質に絞り込むことが一般的である.この過程で切り捨てられるのは,発現の変化が微弱なタンパク質や再現性よく検出されなかったタンパク質である.このようなデータ解析の段階におけるバイアスも手伝ってか,これまでのプロテオーム解析ではがん代謝の全体像は不鮮明であった.
がんの代謝ランドスケープを明らかにするためには何が必要なのだろうか? まず,従来のプロテオーム解析に比べより正確かつ確実性の高いタンパク質の定量法が必要であり,これはすでに述べたように,iMPAQTの導入により達成される.もうひとつ重要な点は,がんに特有の代謝の変化を記述するためには必ず,比較すべき正常な代謝をもつ対照の検体が必要なことである.しかしながら,これまでのがんのプロテオーム解析の多くは臨床検体やがん細胞株を対象としており,的確な対照は存在しなかった.臨床検体ではがん部と非がん部との比較が頻繁になされるが,がんが組織のどのような細胞に由来しているか不明なことも多く,厳密な比較は不可能である.がん細胞株の場合も同様であり,対応する正常な組織に由来する培養細胞を対照として用いるしかない45).
筆者らは,ヒトの正常な細胞株に各種のがん原遺伝子を発現させることにより人工的にがんの状態を再現したモデルシステムを構築し,がん化の前後の代謝経路の違いを詳細に調べた31,46).ヒトの正常な線維芽細胞であるTIG-3細胞にテロメラーゼの構成タンパク質であるTERTおよびSV40の初期遺伝子領域(ラージT抗原およびスモールT抗原をコード)を発現させたTS細胞を得た.さらに,TS細胞にc-Myc遺伝子を発現させたTSM細胞,および,TS細胞に活性化H-RasG12V遺伝子を発現させたTSR細胞を得た.増殖の速度やコロニー形成実験から,TS細胞は増殖の速度は上昇しているがコロニー形成能をもたない不死化細胞であり,TSM細胞およびTSR細胞はさらに速い増殖とコロニー形成能をもつがん化細胞であることが判明した.
正常な細胞には低酸素の環境に応答した解糖系の促進が認められるが,TSM細胞およびTSR細胞においては正常な酸素分圧のもとでも解糖能が高く,がん原遺伝子により誘導したがん状態においてWarburg効果の再現されることが示された31).これらの細胞における代謝酵素の発現を網羅的に計測し比較解析したところ,ヘキソキナーゼ2,グルコース-6-リン酸イソメラーゼ,ホスホフルクトキナーゼM,乳酸デヒドロゲナーゼBなどの解糖系に関連する酵素の発現の亢進が認められ,これらがWarburg効果を生み出していることが推察された31)(図6a).解糖系に関連する多くの酵素にはそれぞれアイソザイムが存在するが,がん原遺伝子の作用はアイソザイムに特異的であり,それぞれのアイソザイムごとに制御や機能が異なることが示唆された.また,解糖系に関連する酵素の変化にくわえ,広範な代謝酵素に発現量の変化が認められた.たとえば,一炭素回路を構成するセリン-グリシン合成経路,葉酸回路,メチオニン回路にかかわる酵素のほとんどがTSM細胞において協調的な発現の亢進が認められた(図6b).このように,がん原遺伝子は全体的な代謝経路の再構築をひき起こし増殖に適した代謝状態をつくりだしていることがプロテオームのレベルで実証された.
iMPAQTにより,1マシンあたり1時間で数百タンパク質というスループットで経路の全体を対象としたタンパク質の一斉定量が可能になった.これまで,個々のタンパク質の単位で実施されてきた生命現象の理解のための仮説の検証が飛躍的に加速するとともに,近傍の経路へと解析の対象を広げることも可能になる.つまり,探索性と検証性とをあわせもつ新次元のタンパク質の研究法が実現しつつある.iMPAQTを用いれば,多様な条件において任意のタンパク質のセットの変化を連続的に追跡することが可能である.これは,ようやくプロテオームの研究が生命システムの動作原理の解明に対して第一歩を踏みだしたことを意味する.
現在,多数のオミクスの階層を横断的に計測するマルチオミクスや,それらを統合して理解するトランスオミクスの重要性が認識されつつある.たとえば,ゲノムの変化と遺伝子の発現量やタンパク質の発現量との関係性をもとめる研究や47),遺伝子の発現量とタンパク質の発現量とのあいだの関係性を半減期モデルで説明する試みなどがなされている48).このような動向のなかで,プロテオームの階層はほかのオミクスの計測の技術に比べて大きく出遅れた感があり,プロテオームの情報が不在のトランスオミクス解析が散見される残念な状況にある.筆者らは,iMPAQTを含めた大規模なターゲットプロテオミクス技術の成熟がこのような状況に歯止めをかけ,真の多階層オミクスデータの取得が可能になると信じている.このような真の多階層オミクスデータの計測によりこれまで知り得なかった生命システムの動作原理がひも解かれ,新たな生命科学の研究の潮流の生まれることが期待される.そのためには,精度の高いオミクスデータとそれを解釈するための新たな枠組みが必要であり,実験系の研究者と情報系あるいは数理系の研究者がより密に連携した研究体制の構築が急務である.
略歴:1998年 福岡大学大学院理学研究科 修了,同年 九州大学生体防御医学研究所 博士研究員,2007年 同 助教を経て,2009年より同 准教授.
研究テーマ:プロテオミクスに関する技術の開発および応用.
抱負:つねに新しい技術を開発しながら,自らがつくった技術を駆使して新しい生命科学の研究のスタイルを築きたい.生命システムらしさが生まれる原理をタンパク質のダイナミクスから明らかにしたい.
中山 敬一(Keiichi I. Nakayama)
略歴:1990年 順天堂大学大学院医学研究科 修了,米国Washington大学 博士研究員,1995年 日本ロシュ研究所 主幹研究員を経て,1996年より九州大学生体防御医学研究所 教授.
研究テーマ:プロテオミクスを利用した医学の研究.
抱負:仮説駆動型の研究とデータ駆動型の研究とを組み合わせ,真のトランスオミクス研究を用いて生命の駆動の原理や疾患の発症の分子機構を理解したい.とくに,罹患率が高く社会的にも大きな問題であるがんおよび精神疾患に対し,根本的な治療法の開発につなげたい.
研究室URL:http://www.bioreg.kyushu-u.ac.jp/saibou/index.html
© 2017 松本雅記・中山敬一 Licensed under CC 表示 2.1 日本
(九州大学生体防御医学研究所 プロテオミクス分野)
email:松本雅記,中山敬一
領域融合レビュー, 6, e002 (2017) DOI: 10.7875/leading.author.6.e002
Masaki Matsumoto & Keiichi I. Nakayama: Life science relying on accurate and reproducible quantitative proteomics.
要 約
近年の質量分析計のいちじるしい高性能化および種々の定量技術の開発によりプロテオーム解析の技術は成熟し,いちどに数千のタンパク質の同定および定量が可能になり生命科学のさまざまな領域の発展に大いに貢献している.その一方で,現在,普及している手法は定量性や再現性の問題に直面しており新たな方法論の構築が望まれてきた.近年,従来のプロテオーム解析法の問題点を解消できる手法として多重反応モニタリング法などの技術を用いたターゲットプロテオミクスが確立され,任意のタンパク質のハイスループットかつ正確な定量が可能になった.しかしながら,多重反応モニタリング法を実施するために必要な情報リソースの不足およびその測定メソッドの構築の煩雑さから,広く普及するにはいたっていない.最近,筆者らを含むいくつかの研究グループから,大規模なターゲットプロテオミクスを容易に可能にする技術開発が報告され,ようやく精密な定量プロテオミクスにもとづく生命科学の研究がスタートしつつある.このレビューにおいては,質量分析計を用いたプロテオーム解析の原理および特徴を説明するとともに,最新のターゲットプロテオミクスの動向を筆者らによる最近の取り組みもまじえて解説し,さらに,がん代謝の研究への応用について紹介する.
はじめに
ヒトなど主要な生物種のゲノム情報の解読を背景として,さまざまなオミクス解析の技術が発展し,網羅的な分子計測データから生命現象をシステムとして理解する試みがなされている.とくに,次世代シークエンス技術の登場はハイスループットかつ網羅性の高い塩基配列の情報の取得を可能にし,さまざまな生命現象や疾患とゲノム,エピゲノム,トランスクリプトームとの関係性が明らかにされている.その一方で,これら遺伝情報と生命現象とをつなぐ原理や法則の蓄積は乏しく,いまだ生命システムへの本質的な理解からはほど遠い状況にある.たとえば,がんはさまざまなゲノムの変異を原因として生じるが,これらの変異がいかにしてがん細胞のさまざまな特徴を生み出すのかについてはほとんどわかっていない.すなわち,生命システムの表現型,たとえば,がんの増殖の速度や運動能などと,さまざまな核酸分子のプロファイルとのあいだには巨大なブラックボックスが存在する.このようなブラックボックスの解消のためには,表現型と相関する分子シグネチャーの同定が必須である.なかでも,生命現象の直接的な担い手であるタンパク質の量的あるいは質的な変化をグローバルにとらえるプロテオーム解析の重要性は明白であり,ゲノム情報の整備および質量分析計の高性能化を追い風に大きな期待がよせられてきた1).
現在,液体クロマトグラフィー(LC)とタンデム質量分析計(MS/MS)とを連結した,いわゆるLC-MS/MSによるプロテオーム解析法がもっとも普及している.LC-MS/MSによるプロテオミクスは,その原理からノンターゲットプロテオミクスとターゲットプロテオミクスの2つに大別される2)(図1).また,タンパク質を酵素により消化して得られるペプチドを解析するボトムアッププロテオミクスと,タンパク質を消化せずにそのまま解析するトップダウンプロテオミクスとに分類することもできるが(図1),このレビューでは,現時点においてより実用的なボトムアッププロテオミクスについてのみ記述する.なお,ボトムアッププロテオミクスは,より慣用的かつ普及しているショットガンプロテオミクスとよぶことにする.
このレビューにおいては,ノンターゲットプロテオミクスおよびターゲットプロテオミクスの原理およびその特徴について解説するとともに,近年,急速に進んだターゲットプロテオミクスの大規模化の動向,さらに,がん代謝の研究への応用について紹介する.
1.探索的な研究のためのノンターゲットプロテオミクス
現在,ショットガンプロテオミクスのなかでもっとも普及しているのは,データ依存的解析(data-dependent acquisition:DDA)法によるノンターゲットプロテオミクスである.通常,複雑なペプチドの混合物は液体クロマトグラフィーにより分離してもなお多数のペプチドが同時に質量分析計に導入されるため,ひとつのMSスペクトルから多数のペプチドに由来するシグナルが検出される.データ依存的解析法においては,MSスペクトルから検出される多数のシグナルのうち強度の強いものを親イオンとして自動的に選択し,衝突誘起解離(collision-induced dissociation:CID)法によりMS/MSスペクトルを取得する(図2).得られたMS/MSスペクトルはペプチドの配列を反映する断片イオンの質量の情報を含む.通常,タンパク質はトリプシンなどのアミノ酸配列に特異的な酵素により消化されるため,親イオンの質量をもとに,タンパク質の配列のデータから候補となるペプチドの配列が絞り込まれる.候補となるペプチドの配列から理論的なMS/MSスペクトルが得られるが,これを実測されたMS/MSスペクトル(ペプチド結合がランダムに切断された場合に生じる断片イオン)と照合し,パターンの一致するペプチドの配列を同定する.ノンターゲットプロテオミクスは興味のある検体に含まれるタンパク質の同定を目的としており,いちどに多数のタンパク質を検出できるため,おもに探索的な研究に利用されている.近年では,ノンターゲットプロテオミクスに定量法を組み合わせることにより,より定量的な解析が可能になっている.定量の技術としては,スペクトルカウント法,標識フリー定量法,in vivo標識法,in vitro標識法があげられる.
スペクトルカウント法(図3a):ノンターゲットプロテオミクスにおいては,おのおののタンパク質において同定されるペプチドの数はタンパク質の存在量におおむね依存する.つまり,同定されたペプチドの数を比較することにより,おのおのの検体のあいだの発現量の比較が可能である.スペクトルカウント法は使用する装置の分解能や質量精度に依存しないため,もっとも汎用性の高い手法である.また,同定されたペプチドの数をタンパク質の分子量や理論ペプチド数で標準化する手法や,これを拡張したexponentially modified protein abundance index(emPAI)などが開発され3),比較定量のみならずタンパク質の存在量の推定にも利用されている.
標識フリー定量法(図3b):個々のペプチドイオンのシグナルからクロマトグラムを得ることによりペプチドの存在量を概算する手法である.特別な前処理が不要であり,あらゆる試料に適用できる手軽な手法である.ただし,特定のペプチドの抽出クロマトグラムを正確に得るためには高分解能かつ高質量精度の質量分析計を必要とする.また,定量性を保証するためには基本的にシングルショット(前分画を施さないLC-MS)での解析が前提であり,比較的発現量の高いタンパク質が解析の対象となる.
in vivo標識法(図3c):培養細胞などを培養する際に安定同位体により標識した物質を取り込ませることで代謝的にタンパク質を標識する.とくに,安定同位体により標識したアミノ酸を用いる方法が普及しており,SILAC(stable isotope labeling with amino acid in cell culture)法とよばれている4).細胞の発現するすべてのタンパク質が安定同位体により標識されるため,試料の調製の初期段階において検体を混合することができ,試料の調製の過程におけるブレを最小限に抑えることが可能である.したがって,オルガネラの画分やアフィニティー精製など複雑な試料の調製を必要とする実験法との相性がよい.
in vitro標識法(図3d):代謝的な標識が適用できない臨床検体などを標識するのに,安定同位体を含む試薬を用いてタンパク質あるいはペプチドを修飾する手法である.個々の検体を異なる質量をもつ試薬により標識するnon-isobaricタグ法5) と,異なる質量をもつリポーター部位とバランサー部位とを組み合わせて同一の質量をもたせたisobaric試薬により標識するisobaricタグ法6) とに分けられる.non-isobaricタグ法においてはおのおのの検体に由来するペプチドが異なる質量をもつ親イオンとして観測され,そのシグナルの強度から比較定量が可能である(質量分析計における定量の原理は,SILAC法と同じ).一方,isobaricタグ法においては標識ののちのペプチドの質量はまったく同一となり,MSスペクトルにおいては単一のピークとして検出される.このペプチドは衝突誘起解離法によりリポーターとバランサーとのあいだが切断されるため,MS/MSスペクトルの低質量の領域に質量が1 Daずつ異なるリポーターイオンが検出され,これらのシグナルの強度がそのペプチドの試料のあいだの相対量比を示す.
2.ノンターゲットプロテオミクスの生命科学への応用
各種の定量の技術の普及と近年の質量分析計の急速な性能の向上により,ノンターゲットプロテオミクスにより大腸菌など比較的単純なプロテオームに含まれるほとんどのタンパク質の検出が可能になった7).さらに,ヒトやマウスなどより複雑なプロテオームについても,LC-MS/MS解析のまえに分画を施すなどのくふうにより数千から1万程度のタンパク質の同定が可能である8,9).ヒトの培養細胞においてmRNAとして発現の確認される遺伝子は通常10,000~12,000であることを考えると8),ほぼ網羅的なプロテオーム発現の解析が可能になっている.このような技術的な成熟を反映してか,近年,2つの研究グループより,ヒトの全プロテオームのドラフトが報告された10,11).いずれの論文も,公共リポジトリに登録された質量分析計のデータに,おのおのの研究グループにおいて新たに取得したデータをくわえ,統一的な解析パイプラインにより処理することで結果の統合を図っている.それぞれ,18,097種類および17,294種類のタンパク質を同定しており,ヒトのゲノムに存在するタンパク質をコードする遺伝子の約9割が実在のタンパク質として発現が確認されたことになる.さまざまな計測条件において取得されたデータが混在しているため,スペクトルカウント法や標識フリー定量法を用いてタンパク質の発現量を大まかに推定するにとどままっているが,ヒトのプロテオーム発現の全体像が得られた意義は大きい.その一方で,これらの報告は,データ依存的解析法による多数のデータを集積させたことで多くの偽陽性ヒットを含む可能性が指摘されており12),多数のタンパク質の存在がいまだに実験的に証明されていない可能性がある.このようなデータ依存的解析法による多数のデータを統合する際の新たな枠組みの開発などが今後の重要な課題であろう.たとえば,2015年より,わが国初の公共プロテオーム情報データベースであるjPOST(Japan Proteome Standard Repository/Database)の開発がはじまり(URL:http://jpostdb.org/),今後,より高質かつ信頼性の高いプロテオーム情報の公開が期待されている.
このように,データ依存的解析法によるノンターゲットプロテオミクスが進む一方で,技術的な制約による問題点もうきぼりになっている.データ依存的解析法により同定されるタンパク質の数は取得したMS/MSスペクトルの数に依存するため,網羅性をあげるには必然的に長い分析時間を要する.すなわち,網羅性とスループットには完全にトレードオフの関係がある.最近,普及しているフーリエ変換型ハイブリッド質量分析計を用いてデータ依存的解析法により2時間程度のデータの取得を実施すると約2000種類のタンパク質が同定されるが,このなかには細胞骨格タンパク質やリボソームタンパク質など発現量が高いものが多く,シグナル伝達タンパク質や転写因子などはまばらである.これは,データ依存的解析法によるMS/MSスペクトルの取得がタンパク質の存在量の多い順に実施されることに起因しており,発現量の低いタンパク質の検出をもとめる場合には1試料あたり4時間から6時間以上の分析時間を要する.つまり,データ依存的解析法では高スループットかつ発現量の低いタンパク質にアクセスが可能な解析を実施することは困難である.さらに,複雑な試料においてデータ依存的解析法を実施した場合,MS/MSスペクトルの取得がランダムになされるうえ,すべてのMS/MSスペクトルが高品質で得られるわけでないため,測定ごとに同定されるペプチドのリストが異なり,再現性の面においても大きな問題となる.
3.ターゲットプロテオミクスの原理
データ依存的解析法における問題や限界を相補する手法として,ターゲットプロテオミクスがきわめて有効である13-15).ターゲットプロテオミクスでは三連四重極型質量分析計を用いた多重反応モニタリング(multiple reaction monitoring:MRM)法(あるいは,選択反応モニタリング法,selected reaction monitoring:SRM法)を用いるのが一般的である.多重反応モニタリング法では,親イオンを通すQ1フィルターと開裂ののちの断片イオンを通すQ3フィルターの組合せ設定(MRMトランジッション)により,特定のイオンを特異的に検出かつ定量する(図4a).通常,ひとつのペプチド(Q1)に対して複数の断片(Q3)のMRMトランジッションを設定するが,これらはクロマトグラムにおいて完全に共溶出するピークとして検出される(図4b).実際の測定においては近傍にノイズのシグナルが生じることも多いが,これら共溶出を指標に目的とするペプチドのピークを同定できる.多重反応モニタリング法はいちどに計測できるペプチドの数に制限があるが,広いダイナミックレンジと高い選択性をもつことから,比較的発現量の低いタンパク質でも事前の分画なしに再現性よく定量することが可能である.また,安定同位体による標識を施した濃度が既知の内部標準を添加することにより正確な絶対定量も可能である.たとえば,出芽酵母において細胞あたり数十~106分子という広範な発現量のレンジに属する100種類のタンパク質を対象として絶対量の計測が実施されている16).このような高速性や定量性などの多重反応モニタリング法の利点は,ノンターゲットプロテオミクスにおいて見い出された興味あるタンパク質の評価の手段としてきわめて重要であり,実際,生体試料におけるバイオマーカーの探索の検証などに活用されている.
最近では,観測されるすべての(あるいは,一定の質量のレンジの)イオンをまとめてMS/MSスペクトルを取得するデータ非依存的解析(data-independent acquisition:DIA)法とよばれる方法が開発され普及しはじめている17).データ非依存的解析法により取得したデータのなかには,理論的にはすべての親イオンに由来する混合MS/MSスペクトルがクロマトグラムの溶出にしたがい格納されている.したがって,任意のペプチドに関するプロダクトイオンの抽出イオンクロマトグラム(すなわち,高分解能の擬似MRMクロマトグラム)を,事前に特定のペプチドに対する測定メソッドを組むことなく,データを取得したのちに表示することが可能である.しかしながら,データ非依存的解析法は多重反応モニタリング法による解析に比べ感度面で若干劣る状況であり,発現量の低いタンパク質の定量にはより高感度な装置の開発が望まれる.
4.大規模なターゲットプロテオミクス
近年では,質量分析計の高速化やスケジュール化(液体クロマトグラフィーにおける保持時間の指定)の発達により,より多成分に対する多重反応モニタリング法による解析や数百のペプチドの一斉定量が可能になっている.これまで,生命科学の研究において,免疫ブロット法などを用いてさまざまな条件におけるタンパク質の発現の挙動を計測することにより“タンパク質Xが生命現象Yにかかわる”という仮説が検証されてきたが,これを多成分で実施できる意義はきわめて大きい.多くのタンパク質はさまざまな基準により機能単位にカテゴライズされる.たとえば,解糖系やTCA回路などの代謝モジュールはほとんどが100種類以下のタンパク質から構成される.主要なシグナル伝達経路も同様におおむね100種類のタンパク質から構成される.また,タンパク質の機能単位でみても,多くの場合は数十~数百のタンパク質のメンバーから構成されるファミリーである.したがって,数十~数百のタンパク質を確実に一斉定量できれば,経路や機能単位の情報の取得が可能になる.
近年,多重反応モニタリング法を基盤としたターゲットプロテオミクスを大規模な解析に利用しようとする機運が高まり,そのためのインフラの構築が世界中で進められてきた18-20).多重反応モニタリング法においては,事前にタンパク質に特異的かつ高感度な対象ペプチド(proteotypic peptide:PTP)の選定,および,おのおののペプチドに対するMRMアッセイ(MRMトランジッションの組合せ)の作成および検証が必要である(図4b).PTPはおのおのの研究者が自身で同定したタンパク質のペプチドから選ぶことも可能であるが,現在では,データ依存的解析法により世界中で取得されたデータが各種のリポジトリサイトを介して集約され,高頻度に同定されるペプチドがその候補として利用されており,多重反応モニタリング法の普及を促進する重要な基盤となっている21).しかしながら,これらの情報はあくまでPTPの候補であり,実際には,これらのなかからより感度の高いペプチドの選定や測定に必要な情報(どの断片イオンを使うか,液体クロマトグラフィーにおける保持時間の情報など)を合成ペプチドなどの標品を用いて取得し,MRMメソッドの有効性を評価する必要があり,これが実際に多重反応モニタリング法を大規模に実施しようとした際に大きな障壁となっている.
MRMメソッドの構築の迅速化のため,ハイスループットなペプチド合成法により作製したペプチドライブラリーを用いた大規模なMRMメソッドの評価が実施されている.このような試みは,比較的ゲノムサイズの小さい出芽酵母や結核菌などから実施され,その戦略が確立されてきた22-24).最近,同様の手法によりヒトのプロテオームを対象とした網羅的なMRMメソッドが構築され,データベースが公開されている25)(human SRMAtlas resource,URL:http://www.srmatlas.org).このデータベースにおいては,実試料から同定されたペプチドにくわえ,バイオインフォマティクスによる予測により16万以上のPTPの候補の合成ペプチドを作製しMRMメソッドが精査されている.さらに最近では,大規模な合成ペプチドライブラリーの構築をめざしたプロジェクトProteomeTools(URL:http://www.proteometools.org)の一部(35万以上のペプチド)に関する報告がなされた26).ここでは,さまざまな機種あるいはイオンの開裂法によりMS/MSスペクトルを取得しているが,多重反応モニタリング法による評価は実施していない.その一方で,将来的に,トリプシン以外の酵素により生じるペプチドや修飾ペプチドまで拡張することを明言しており,将来的に,より網羅性の高いスペクトルライブラリーの充実がみこまれる.
このように,合成ペプチドを利用したMRMメソッドの構築が進められてきた一方で,合成ペプチドを用いるターゲットプロテオミクスに対する警鐘も鳴らされている27).おのおののタンパク質からの酵素消化によるペプチドの切り出しの効率が必ずしも完全ではない点や28),予測にもとづき合成された候補のペプチドが必ずしも高感度なペプチドである保証がないことなどから,MRMメソッドの構築に組換えタンパク質の利用が提案されてきた27,29).しかしながら,一般の研究室では組換えタンパク質の作製や精製は個々に実施されており,時間と労力を要するものであった.そのため,網羅的な組換えタンパク質を用いた大規模なMRMメソッドの構築やその評価は実施されていなかった.
筆者らは,Gatewayシステムに組み込まれたヒトの完全長cDNAライブラリーを用いた大規模な組換えタンパク質リソースであるin vitro proteomeに着目し30).質量分析に適した試料の調製法を確立した.最終的に,18,000種類以上のゲノムワイドな組換えタンパク質に対するMS/MSスペクトルの取得(200,000以上のペプチド)に成功した31).さらに,これらのMS/MSスペクトルの情報を利用して網羅的にMRMメソッドを構築し,実際の測定にもとづき精査することにより,ゲノムワイドなターゲットプロテオミクスを実施するための基盤情報の取得を完了した.さらに,組換えタンパク質を酵素消化した産物にはmTRAQ法による安定同位体の質量タグを付加しており,これらのリソースはMRMメソッドの構築のための情報源のみではなく,タンパク質の絶対定量の際の内部標準としても利用が可能である31).また,多重反応モニタリング法による計測に必要な情報を格納したデータベースと定量解析のためのツールも独自に作成しており,これらを含めたプラットフォームをiMPAQT(in vitro proteome-assisted MRM for protein absolute quantification)と名づけて公開している(URL:http://impaqt.jpost.org,図5).このデータベースでは,さまざまな分子機能,関与する生命現象,生化学的な経路にもとづいて任意のタンパク質を検索し,対応するMRMメソッドを取得することが可能である.
実際に,このデータベースを利用したところ,代謝経路に関連する約1000種類の酵素に関するMRMメソッドの設計および最適化は数日で完了し実試料の分析に進むことができた.そこで,すべての代謝酵素を対象とした定量代謝マップの作成に取り組み,ヒトの正常な線維芽細胞において発現する約650種類の代謝酵素の発現絶対量の計測に成功した31).その結果,解糖系の後半の反応にかかわるグリセルアルデヒド-3-リン酸デヒドロゲナーゼや乳酸デヒドロゲナーゼAなどはきわめて発現が高いのに対し,解糖系の律速段階であるヘキソキナーゼやホスホフルクトキナーゼは発現量の低いことが判明した.これらの酵素の発現量を低く抑えることが反応律速をつくりだすのに寄与する可能性が示唆された.このように,代謝経路の定量的な特徴は個々の酵素の機能や経路における位置を反映するのかもしれない.
5.がんにおける代謝の全体像
古くから,がんと代謝のあいだには密接な関係性が知られている.その代表的なものが,好気的な条件における解糖系の亢進,Warburg効果である32,33).このがんにおける解糖系の亢進は非代謝性のグルコースアナログを利用した陽電子断層撮像法(positron emission tomography:PET)としてがんの画像診断に応用されていることからも,多くのがんにおいて普遍的であることが広く認知されている34).しかしながら,どのような機構によりがんに特有の代謝状態が生じるのかについては不明な点も多い.近年,c-Myc遺伝子,HIF1遺伝子,p53遺伝子などがんに関連する遺伝子の変異や発現量の変化がさまざまな代謝酵素の発現に直接的に作用し,代謝経路を変化させていることが明らかにされ注目されている32,35,36).たとえば,c-Myc遺伝子は乳酸デヒドロゲナーゼA,ヘキソキナーゼ2,ピルビン酸キナーゼM2など解糖系に関連する多くの酵素の発現を上昇させる36).低酸素に応答性の転写因子HIF1の活性化は解糖系に関連するほとんどの酵素の発現を上昇させる.また,がん抑制遺伝子であるp53遺伝子は,グルコーストランスポーターやヘキソキナーゼ2など解糖系に関連する酵素の発現の抑制や,解糖系を負に制御するTIGARの発現の亢進により解糖系を抑制する37).さらに,p53はグルコース-6-リン酸脱水素酵素の発現の抑制を介したペントースリン酸回路の抑制やシトクロムcオキシダーゼ合成酵素2の発現の亢進による酸化的リン酸化の促進なども担う37).したがって,p53の代謝制御能の喪失は相対的に解糖系の活性化をひき起こすと考えられている.実際のがんにおいては,これらがんに関連する遺伝子の変化は複合的であり,その結果として生じる代謝経路の再編成はきわめて広範囲かつ複雑である可能性が高い.
代謝は多数の酵素の織りなす生化学反応の集積であり,細胞システムは内外の環境の摂動に応じて代謝経路をダイナミックに再編することによりさまざまな環境に適応する.がんに関連する遺伝子が代謝に直接的に影響をあたえることにくわえ,がん細胞は生体において微小環境の要因の影響も大いにうけると考えられるため,がん化にともなう代謝システムの変化はきわめて広範囲かつ多様な様相を示すと考えられている.代謝システムの状態を記述するには細胞における代謝酵素の活性を測定することがもっとも直接的であるが,現時点において,代謝酵素の活性を網羅的に計測する手立ては存在しないため,代謝酵素の発現量から酵素活性を推定するのが現実的である.したがって,がん代謝の研究への発現プロテオーム解析の適用の重要性は早くから認識されており,実際に,さまざまな研究が展開されてきた38).たとえば,初期のプロテオーム研究においては,2次元電気泳動を用いてさまざまながんの臨床検体を対象としたタンパク質の発現量の比較がなされており,がんにおいて発現の上昇するタンパク質のスポットとして解糖系に関連する多くの酵素が同定されている38-40).そののち,ショットガンプロテオミクスが普及し,がん細胞においてより広範な代謝酵素の発現量の変化が生じていることが示された41,42).近年では,大規模なプロテオームデータの取得とほかのオミクスデータとの統合により,がんにより生じるゲノムの変化とプロテオームとのあいだの関連に着目した研究もなされている43-45).
このようにがんのプロテオーム解析が世界中で進むなか,がん代謝の理解はどのくらい進んだのだろうか? 確かに,いちどに数千のタンパク質を同定し比較できるようにはなったが,さきに述べたように,この手法は再現性や定量性においていまだ問題点も多い.とくに,情報量が膨大かつノイズの多いタンパク質のリストのデータ解析においては,多くのデータを切り捨て特徴的な挙動を示すタンパク質に絞り込むことが一般的である.この過程で切り捨てられるのは,発現の変化が微弱なタンパク質や再現性よく検出されなかったタンパク質である.このようなデータ解析の段階におけるバイアスも手伝ってか,これまでのプロテオーム解析ではがん代謝の全体像は不鮮明であった.
がんの代謝ランドスケープを明らかにするためには何が必要なのだろうか? まず,従来のプロテオーム解析に比べより正確かつ確実性の高いタンパク質の定量法が必要であり,これはすでに述べたように,iMPAQTの導入により達成される.もうひとつ重要な点は,がんに特有の代謝の変化を記述するためには必ず,比較すべき正常な代謝をもつ対照の検体が必要なことである.しかしながら,これまでのがんのプロテオーム解析の多くは臨床検体やがん細胞株を対象としており,的確な対照は存在しなかった.臨床検体ではがん部と非がん部との比較が頻繁になされるが,がんが組織のどのような細胞に由来しているか不明なことも多く,厳密な比較は不可能である.がん細胞株の場合も同様であり,対応する正常な組織に由来する培養細胞を対照として用いるしかない45).
筆者らは,ヒトの正常な細胞株に各種のがん原遺伝子を発現させることにより人工的にがんの状態を再現したモデルシステムを構築し,がん化の前後の代謝経路の違いを詳細に調べた31,46).ヒトの正常な線維芽細胞であるTIG-3細胞にテロメラーゼの構成タンパク質であるTERTおよびSV40の初期遺伝子領域(ラージT抗原およびスモールT抗原をコード)を発現させたTS細胞を得た.さらに,TS細胞にc-Myc遺伝子を発現させたTSM細胞,および,TS細胞に活性化H-RasG12V遺伝子を発現させたTSR細胞を得た.増殖の速度やコロニー形成実験から,TS細胞は増殖の速度は上昇しているがコロニー形成能をもたない不死化細胞であり,TSM細胞およびTSR細胞はさらに速い増殖とコロニー形成能をもつがん化細胞であることが判明した.
正常な細胞には低酸素の環境に応答した解糖系の促進が認められるが,TSM細胞およびTSR細胞においては正常な酸素分圧のもとでも解糖能が高く,がん原遺伝子により誘導したがん状態においてWarburg効果の再現されることが示された31).これらの細胞における代謝酵素の発現を網羅的に計測し比較解析したところ,ヘキソキナーゼ2,グルコース-6-リン酸イソメラーゼ,ホスホフルクトキナーゼM,乳酸デヒドロゲナーゼBなどの解糖系に関連する酵素の発現の亢進が認められ,これらがWarburg効果を生み出していることが推察された31)(図6a).解糖系に関連する多くの酵素にはそれぞれアイソザイムが存在するが,がん原遺伝子の作用はアイソザイムに特異的であり,それぞれのアイソザイムごとに制御や機能が異なることが示唆された.また,解糖系に関連する酵素の変化にくわえ,広範な代謝酵素に発現量の変化が認められた.たとえば,一炭素回路を構成するセリン-グリシン合成経路,葉酸回路,メチオニン回路にかかわる酵素のほとんどがTSM細胞において協調的な発現の亢進が認められた(図6b).このように,がん原遺伝子は全体的な代謝経路の再構築をひき起こし増殖に適した代謝状態をつくりだしていることがプロテオームのレベルで実証された.
おわりに
iMPAQTにより,1マシンあたり1時間で数百タンパク質というスループットで経路の全体を対象としたタンパク質の一斉定量が可能になった.これまで,個々のタンパク質の単位で実施されてきた生命現象の理解のための仮説の検証が飛躍的に加速するとともに,近傍の経路へと解析の対象を広げることも可能になる.つまり,探索性と検証性とをあわせもつ新次元のタンパク質の研究法が実現しつつある.iMPAQTを用いれば,多様な条件において任意のタンパク質のセットの変化を連続的に追跡することが可能である.これは,ようやくプロテオームの研究が生命システムの動作原理の解明に対して第一歩を踏みだしたことを意味する.
現在,多数のオミクスの階層を横断的に計測するマルチオミクスや,それらを統合して理解するトランスオミクスの重要性が認識されつつある.たとえば,ゲノムの変化と遺伝子の発現量やタンパク質の発現量との関係性をもとめる研究や47),遺伝子の発現量とタンパク質の発現量とのあいだの関係性を半減期モデルで説明する試みなどがなされている48).このような動向のなかで,プロテオームの階層はほかのオミクスの計測の技術に比べて大きく出遅れた感があり,プロテオームの情報が不在のトランスオミクス解析が散見される残念な状況にある.筆者らは,iMPAQTを含めた大規模なターゲットプロテオミクス技術の成熟がこのような状況に歯止めをかけ,真の多階層オミクスデータの取得が可能になると信じている.このような真の多階層オミクスデータの計測によりこれまで知り得なかった生命システムの動作原理がひも解かれ,新たな生命科学の研究の潮流の生まれることが期待される.そのためには,精度の高いオミクスデータとそれを解釈するための新たな枠組みが必要であり,実験系の研究者と情報系あるいは数理系の研究者がより密に連携した研究体制の構築が急務である.
文 献
- Pandey, A. & Mann, M.: Proteomics to study genes and genomes. Nature, 405, 837-846 (2000)[PubMed]
- Aebersold, R. & Mann, M.: Mass-spectrometric exploration of proteome structure and function. Nature, 537, 347-355 (2016)[PubMed]
- Ishihama, Y., Oda, Y., Tabata, T. et al.: Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics, 4, 1265-1272 (2005)[PubMed]
- Ong, S. E., Blagoev, B., Kratchmarova, I. et al.: Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics, 1, 376-386 (2002)[PubMed]
- Gygi, S. P., Rist, B., Gerber, S. A. et al.: Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol., 17, 994-999 (1999)[PubMed]
- Ross, P. L., Huang, Y. L. N., Marchese, J. N. et al.: Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics, 3, 1154-1169 (2004)[PubMed]
- Iwasaki, M., Miwa, S., Ikegami, T. et al.: One-dimensional capillary liquid chromatographic separation coupled with tandem mass spectrometry unveils the Escherichia coli proteome on a microarray scale. Anal. Chem., 82, 2616-2620 (2010)[PubMed]
- Nagaraj, N., Wisniewski, J. R., Geiger, T. et al.: Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol., 7, 548 (2011)[PubMed]
- Beck, M., Schmidt, A., Malmstroem, J. et al.: The quantitative proteome of a human cell line. Mol. Syst. Biol., 7, 549 (2011)[PubMed]
- Wilhelm, M., Schlegl, J., Hahne, H. et al.: Mass-spectrometry-based draft of the human proteome. Nature, 509, 582-587 (2014)[PubMed]
- Kim, M. S., Pinto, S. M., Getnet, D. et al.: A draft map of the human proteome. Nature, 509, 575-581 (2014)[PubMed]
- Ezkurdia, I., Vazquez, J., Valencia, A. et al.: Analyzing the first drafts of the human proteome. J. Proteome Res., 13, 3854-3855 (2014)[PubMed]
- Maiolica, A., Junger, M. A., Ezkurdia, I. et al.: Targeted proteome investigation via selected reaction monitoring mass spectrometry. J. Proteomics, 75, 3495-3513 (2012)[PubMed]
- Carapito, C. & Aebersold, R.: Targeted proteomics. Proteomics, 12, 1073 (2012)[PubMed]
- Marx, V.: Targeted proteomics. Nat. Methods, 10, 19-22 (2013)[PubMed]
- Picotti, P., Bodenmiller, B., Mueller, L. N. et al.: Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell, 138, 795-806 (2009)[PubMed]
- Gillet, L. C., Navarro, P., Tate, S. et al.: Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics, 11, O111.016717 (2012)[PubMed]
- Picotti, P., Rinner, O., Stallmach, R. et al.: High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods, 7, 43-46 (2010)[PubMed]
- MacLean, B., Tomazela, D. M., Shulman, N. et al.: Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics, 26, 966-968 (2010)[PubMed]
- Sharma, V., Eckels, J., Taylor, G. K. et al.: Panorama: a targeted proteomics knowledge base. J. Proteome Res., 13, 4205-4210 (2014)[PubMed]
- Deutsch, E. W., Lam, H. & Aebersold, R.: PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep., 9, 429-434 (2008)[PubMed]
- Picotti, P., Clement-Ziza, M., Lam, H. et al.: A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature, 494, 266-270 (2013)[PubMed]
- Karlsson, C., Malmstrom, L., Aebersold, R. et al.: Proteome-wide selected reaction monitoring assays for the human pathogen Streptococcus pyogenes. Nature Commun, 3, 1301 (2012)[PubMed]
- Schubert, O. T., Mouritsen, J., Ludwig, C. et al.: The Mtb proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe, 13, 602-612 (2013)[PubMed]
- Kusebauch, U., Campbell, D. S., Deutsch, E. W. et al.: Human SRMAtlas: a resource of targeted assays to quantify the complete kuman proteome. Cell, 166, 766-778 (2016)[PubMed]
- Zolg, D. P., Wilhelm, M., Schnatbaum, K. et al.: Building ProteomeTools based on a complete synthetic human proteome. Nat. Methods, 14, 259-262 (2017)[PubMed]
- Abbatiello, S. E., Schilling, B., Mani, D. R. et al.: Large-scale interlaboratory study to develop, analytically validate and apply highly multiplexed, quantitative peptide assays to measure cancer-relevant proteins in plasma. Mol. Cell. Proteomics, 14, 2357-2374 (2015)[PubMed]
- Brownridge, P. & Beynon, R. J.: The importance of the digest: proteolysis and absolute quantification in proteomics. Methods, 54, 351-360 (2011)[PubMed]
- Stergachis, A. B., MacLean, B., Lee, K. et al.: Rapid empirical discovery of optimal peptides for targeted proteomics. Nat. Methods, 8, 1041-1043 (2011)[PubMed]
- Goshima, N., Kawamura, Y., Fukumoto, A. et al.: Human protein factory for converting the transcriptome into an in vitro-expressed proteome. Nat Methods, 5, 1011-1017 (2008)[PubMed]
- Matsumoto, M., Matsuzaki, F., Oshikawa, K. et al.: A large-scale targeted proteomics assay resource based on an in vitro human proteome. Nat Methods, 14, 251-258 (2017)[PubMed]
- Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B.: Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science, 324, 1029-1033 (2009)[PubMed]
- Gatenby, R. A. & Gillies, R. J.: Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer, 4, 891-899 (2004)[PubMed]
- Mankoff, D. A., Eary, J. F., Link, J. M. et al.: Tumor-specific positron emission tomography imaging in patients: [18F]fluorodeoxyglucose and beyond. Clin. Cancer Res., 13, 3460-3469 (2007)[PubMed]
- Stubbs, M. & Griffiths, J. R.: The altered metabolism of tumors: HIF-1 and its role in the Warburg effect. Adv. Enzyme Regul., 50, 44-55 (2010)[PubMed]
- Gordan, J. D., Thompson, C. B. & Simon, M. C.: HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell, 12, 108-113 (2007)[PubMed]
- Bensaad, K. & Vousden, K. H.: p53: new roles in metabolism. Trends Cell Biol., 17, 286-291 (2007)[PubMed]
- Scatena, R., Bottoni, P., Pontoglio, A. et al.: Revisiting the Warburg effect in cancer cells with proteomics. The emergence of new approaches to diagnosis, prognosis and therapy. Proteomics Clin. Appl., 4, 143-158 (2010)[PubMed]
- Bi, X., Lin, Q., Foo, T. W. et al.: Proteomic analysis of colorectal cancer reveals alterations in metabolic pathways mechanism of tumorigenesis. Mol. Cell. Proteomics, 5, 1119-1130 (2006)[PubMed]
- Seike, M., Kondo, T., Fujii, K. et al.: Proteomic signature of human cancer cells. Proteomics, 4, 2776-2788 (2004)[PubMed]
- Sun, Y., Mi, W., Cai, J. et al.: Quantitative proteomic signature of liver cancer cells: tissue transglutaminase 2 could be a novel protein candidate of human hepatocellular carcinoma. J. Proteome Res., 7, 3847-3859 (2008)[PubMed]
- Zhou, W., Capello, M., Fredolini, C. et al.: Proteomic analysis reveals Warburg effect and anomalous metabolism of glutamine in pancreatic cancer cells. J. Proteome Res., 11, 554-563 (2012)[PubMed]
- Gholami, A. M., Hahne, H., Wu, Z. et al.: Global proteome analysis of the NCI-60 cell line panel. Cell Rep., 4, 609-620 (2013)[PubMed]
- Zhang, B., Wang, J., Wang, X. et al.: Proteogenomic characterization of human colon and rectal cancer. Nature, 513, 382-387 (2014)[PubMed]
- Lawrence, R. T., Perez, E. M., Hernandez, D. et al.: The proteomic landscape of triple-negative breast cancer. Cell Rep., 11, 630-644 (2015)[PubMed]
- Akagi, T., Sasai, K. & Hanafusa, H.: Refractory nature of normal human diploid fibroblasts with respect to oncogene-mediated transformation. Proc. Natl. Acad. Sci. USA, 100, 13567-13572 (2003)[PubMed]
- Zhang, H., Liu, T., Zhang, Z. et al.: Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell, 166, 755-765 (2016)[PubMed]
- Schwanhausser, B., Busse, D., Li, N. et al.: Global quantification of mammalian gene expression control. Nature, 473, 337-342 (2011)[PubMed]
著者プロフィール
略歴:1998年 福岡大学大学院理学研究科 修了,同年 九州大学生体防御医学研究所 博士研究員,2007年 同 助教を経て,2009年より同 准教授.
研究テーマ:プロテオミクスに関する技術の開発および応用.
抱負:つねに新しい技術を開発しながら,自らがつくった技術を駆使して新しい生命科学の研究のスタイルを築きたい.生命システムらしさが生まれる原理をタンパク質のダイナミクスから明らかにしたい.
中山 敬一(Keiichi I. Nakayama)
略歴:1990年 順天堂大学大学院医学研究科 修了,米国Washington大学 博士研究員,1995年 日本ロシュ研究所 主幹研究員を経て,1996年より九州大学生体防御医学研究所 教授.
研究テーマ:プロテオミクスを利用した医学の研究.
抱負:仮説駆動型の研究とデータ駆動型の研究とを組み合わせ,真のトランスオミクス研究を用いて生命の駆動の原理や疾患の発症の分子機構を理解したい.とくに,罹患率が高く社会的にも大きな問題であるがんおよび精神疾患に対し,根本的な治療法の開発につなげたい.
研究室URL:http://www.bioreg.kyushu-u.ac.jp/saibou/index.html
© 2017 松本雅記・中山敬一 Licensed under CC 表示 2.1 日本
