集団ゲノミクスからさぐる植物の進化
2017/08/17
土松 隆志
(千葉大学大学院理学研究院 生物学研究部門植物進化ゲノミクス研究室)
email:土松隆志
領域融合レビュー, 6, e006 (2017) DOI: 10.7875/leading.author.6.e006
Takashi Tsuchimatsu: Population genomics reveals evolutionary processes of plant populations.

塩基配列決定技術の飛躍的な発展にともない,近年,全ゲノム情報にもとづく植物の進化の研究がさかんである.とくに,ひとつの種あるいは複数の近縁種にフォーカスして数十から数千におよぶ多数の試料をゲノム配列にもとづき解析する“集団ゲノミクス”の進展がめざましい.集団のゲノムデータを解析することにより,過去から現在にいたる個体数の変動や分化の過程などの歴史を推定できるほか,ゲノムワイド関連解析により自然変異にかかわる遺伝子を効率的に同定することも可能である.このレビューにおいては,モデル植物であるシロイヌナズナの事例を中心に,集団ゲノミクスの手法を解説する.
生物のもつ全遺伝情報であるゲノムには,おもに2つの側面がある.1つ目は,ゲノムは生命活動に必要な情報のコードされた遺伝子の総体であり,さまざまな表現型がかたちづくられる“設計図”としての側面である.そして2つ目は,ゲノムは親から子へと脈々と受け継がれるものであり,生物の進化の痕跡がきざまれた“進化の履歴”としての側面である.現代の進化生物学における主要な課題のひとつは,ゲノムをこの2つの視点から詳細に解剖することにより,生物の集団にみられるさまざまな進化の過程を遺伝子のレベルから復元し予測することである.適応的な表現型はどの遺伝子のどの突然変異により生じてきたのだろうか.まったく新奇な表現型の進化には何個の遺伝子に変異が必要なのだろうか.ある生物種の集団は過去から現在にわたりどのように個体数が変動してきたのだろうか.近縁種どうしの種分化はいつ起こり,そののち,遺伝子の流動はどのくらい起こっているだろうか.塩基配列決定技術の向上にともない,ひとつの種あるいは近縁種における塩基配列の違いを全ゲノムにわたり容易かつ網羅的に把握できるようになったこともあり,近年,これらの問いに答える集団遺伝学の手法も飛躍的に発展している.
このレビューにおいておもにとりあげるのは,植物のひとつの種あるいは複数の近縁種にフォーカスして数十から数千におよぶ多数の試料をゲノム配列にもとづき解析する“集団ゲノミクス”である.はじめに,集団ゲノミクスにおいて扱うデータセットがどのようなものかを解説したうえで,個体数の動態など集団の歴史をどのように推定するか,また,表現型の変異をつかさどる遺伝子をどのように推定するか,という2つの視点から話を進める.
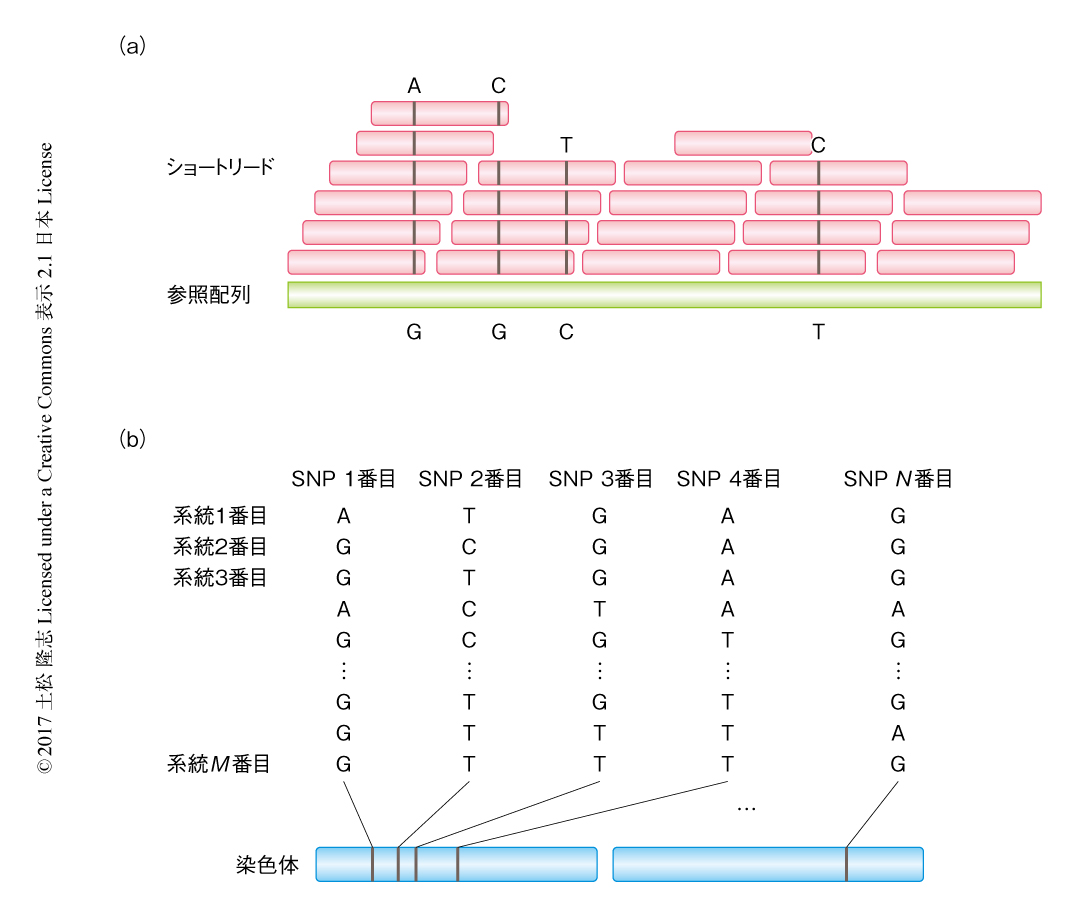
集団ゲノミクスの基本になるデータセットは,ひとつの種あるいは近縁種の数十から数千に及ぶ多数の個体についてゲノムを網羅する大量の遺伝マーカーのデータである.遺伝マーカーは,以前はSanger法などにより得られる断片的な塩基配列にもとづくものが多かったが,最近は次世代シークエンサーを利用し全ゲノム配列を決定するリシークエンス解析によるものが主流になりつつある.リシークエンス解析とは,すでにゲノム配列が解読されている種について,その既知のゲノム配列を参照配列として,次世代シークエンサーにより得られたほかの個体のゲノム配列に由来する短い塩基配列(ショートリード)をマッピングし,参照配列との違いのある部分を検出していく手法である(図1a).参照配列との違いとして得られるものには1塩基多型(single nucleotide polymorphism:SNP)や短い挿入および欠失(indel)などがあるが,もっとも頻繁にみられるSNPを遺伝マーカーとして用いることが多い.標準系統の参照配列に対しショートリードをマッピングし,ゲノムの全体にわたりSNPを抽出する.この作業を多数の個体について行うことにより,SNPの巨大なマトリックスを作成することができる(図1b).これが,さまざまな集団ゲノミクス解析の基礎となる重要なデータセットである.
このSNPのデータセットを用いることにより,たとえば,それぞれの個体が互いにどのくらい近縁かを簡単に計算することができる.ある2つの系統を抽出し,SNPを数えてゲノムの全長で割り算をすると,ゲノムにおいて何%の塩基に置換がみられるかがもとめられる.この値はより近縁であれば小さく,より遠縁であれば大きくなる.すべての個体の組合せについてこの計算を行い平均した値は塩基多様度とよばれ,その種や集団の遺伝的な多様性の基本的な指標になる.塩基多様度は,たとえば,シロイヌナズナで約0.5%,ヒトでは約0.1%であることが知られている.
このような個体のあいだの近縁度の全体像をおおまかに把握するため,ゲノムの類似度を主成分分析などの手法により2次元の平面に投影することがよく行われる.たとえば,ヨーロッパにおけるヒトの集団の遺伝的な類縁度を約3000人,500,568遺伝マーカーの多型データを用いて解析し,主成分分析を用いて可視化した研究がある1).遺伝的な類縁度は集団のあいだの地理的な関係とおおむね対応しており,ヒトの交配や移動が地理的に限定されていることが示唆される.
主成分分析にくわえて,クラスタリング解析も一般的である2).STRUCTUREやその派生のソフトウェアは,集団の全体がN個の分集団に分かれると仮定したとき,おのおのの個体がX番目に属する確率をもとめるものである.もちろん,厳密にN個の分集団に分かれるわけではなく,特定の分集団への帰属がはっきりしない個体もみられることが多い.それでも,対象とする種がおおむね何個の分集団に分かれるのか,この解析からおおまかな目安をつけることは可能である.このように,種や集団が遺伝的に均一に混ざりあっておらず分集団のような構造がみられるとき,これを集団構造とよぶ.主成分分析やクラスタリング解析をとおして,対象とする種の集団構造のパターンを把握することができる.
主成分分析やクラスタリング解析などから明らかにされた集団構造は,その種の進化の歴史を反映している.たとえば,その種が長期にわたり2つの分集団に分かれていたなら,クラスタリング解析により2つのクラスターにはっきり分かれることが予想される.
集団ゲノミクスのデータを用いることにより,集団構造を把握するだけでなく,その背景にある集団の進化の歴史を定量的に推定することができる.ここで,集団の歴史とは具体的に,過去における個体数の増減,種や集団の分化,遺伝子の流動の過程などのことをさす.このような推定のベースにあるのは,コアレセント理論(coalescent theory)とよばれる比較的新しい集団遺伝学の理論である.コアレセント理論とは,集団から得られたDNA配列の系譜を遺伝子系図(gene genealogy)として解析することにより,さまざまな進化的なパラメーターを定量的に推定する集団遺伝学の考え方のひとつである.コアレセント(coalescent)という語は“合着”あるいは“合祖”と訳され,集団のある2つのDNA配列が世代をさかのぼり共通の祖先にいたることをさす(図2).
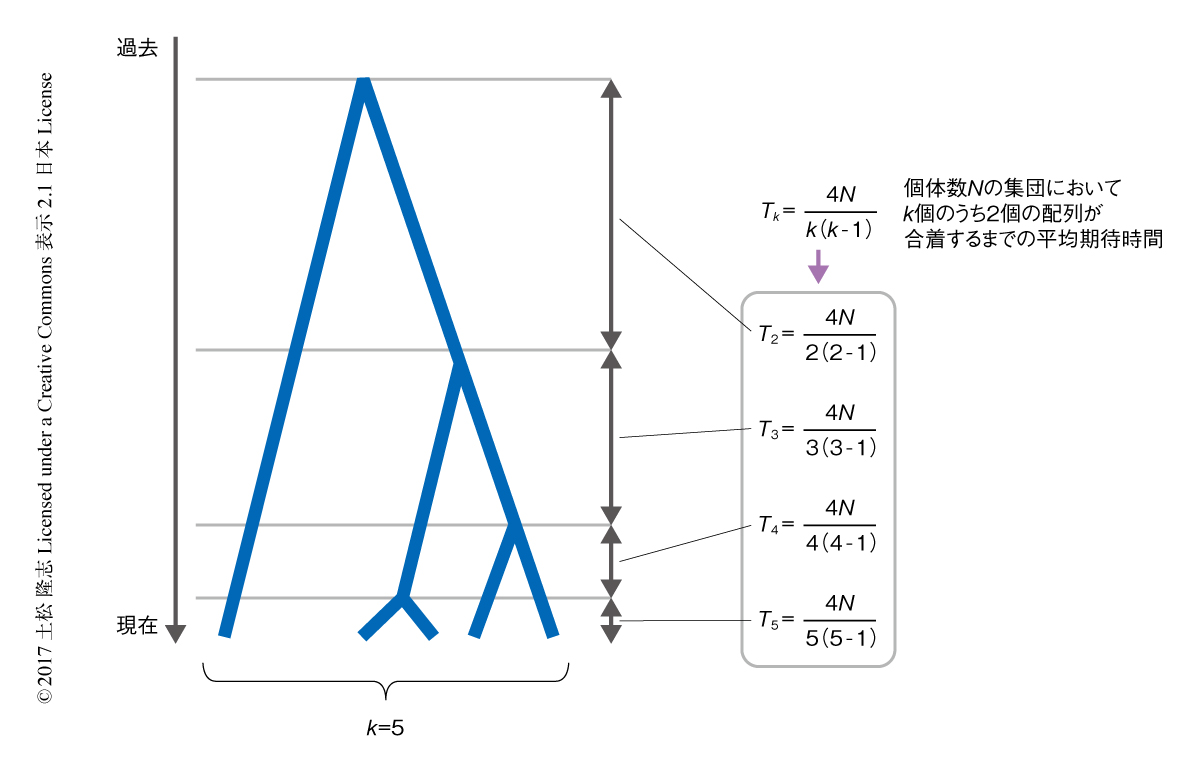
コアレセント理論の核となるコンセプトは,得られたDNA配列の合着が何世代まえにさかのぼるか理論的な期待値がもとめられることである.そして,合着が何世代まえにどれだけ起こったかは,個体数の変動,自然選択,集団構造などの影響をうけて変化する.つまり逆に考えれば,試料として得られたDNA配列の合着がいつどれだけ起こったかを調べれば,過去の集団における個体数の変動などを推定できることになる.おおまかには,集団のサイズが大きいときには合着は相対的に少なく,集団のサイズが小さいときにはより多くの合着が起こる.これは,あるタイミングで個体数が急に1個体に減少した状況を考えるとわかりやすい.すべての試料は最終的に必ずそこで合着するはずである.また,過去のある時期に集団が2つの分集団に分かれていたとすると,その期間の分集団のあいだにおいて合着は少ないことが期待される.
最近,このようなコアレセント理論による個体数の動態の推定を,多数の個体のゲノムワイドな遺伝子多型のデータにもとづき行うアルゴリズムが提案された.MSMC(multiple sequentially Markovian coalescent)とよばれるこの手法は3),2倍体の1個体のゲノム配列から過去の個体数の動態を推定するアルゴリズムPSMC(pairwise sequentially Markovian coalescent)を発展させたものである4).
ここでは,MSMC解析をシロイヌナズナのデータに適用した例を紹介する.シロイヌナズナはおもに中央アジア,ヨーロッパ,北アフリカ,北アメリカに生育する1年草であり,近年,“1001ゲノムプロジェクト”とよばれる1135に及ぶ野生系統のリシークエンス解析が実施されるなど,集団ゲノミクスのモデルとして注目されている5).1001ゲノムプロジェクトのリシークエンスデータをもとにSNPのマトリックスを作成し,すべての系統の組合せについて塩基置換率を計算したところ,おおむね,ピークを約0.005(0.5%)とするきれいな山形になったが,ピークより値の大きい側の裾野にかたよってふくらみがみられた.これは,ほかの大多数の系統とは遠縁な個体がいくつかまぎれていることによると考えられた.調べてみると,この遠縁な系統(ここでは,“遺存系統”とよぶ)は26系統あり,その多くは地中海地方に由来するものだった.MSMC解析を実施したところ,遺存系統と残りの大多数の系統とのあいだにおいては,最終氷期のはじまった7万年前ごろから最終氷期ののちの現在にいたるまで,ほとんど合着がみられないことがわかった5).この結果から,両者のあいだで最終氷期ののちには遺伝的な交流はほとんど起こっていないことが示された.最終氷期におけるシロイヌナズナの分布は南の地中海地方に制限され,いくつかの氷河退避地に集団が分かれていたと考えられている.最終氷期ののち,氷河退避地からふたたび分布が拡大した際に,おそらくはひとつの氷河退避地に由来する系統が圧倒的に頻度を高め,それが現在の多数派の系統となったと予想された.一方,遺伝的に分化した少数派の遺存系統は,いわば氷河期の生き残りとでもよぶべき系統であり,生育地の特性などの表現型に多数派の系統とは異なる特徴がみられると報告されている.
MSMC解析はシロイヌナズナだけでなく,最近,トウモロコシの集団ゲノミクスにも適用されている6).その結果,トウモロコシは近縁の野生種であるテオシンテから1万~1.5万年前に分岐し,その際,集団のサイズが約5%に減少する非常に強いボトルネック効果(びん首効果)のはたらいたことがわかった.そののち,集団のサイズは急速に増加し,現在の栽培トウモロコシの集団ができあがったと推定されている.
コアレセント理論とゲノムデータにもとづき集団の歴史を推定する手法は,MSMC解析のほかにもさまざまな手法が提案されている.たとえば,2つの集団のあいだで遺伝子の流動がどのくらい起こっているか,とくに,その遺伝子の流動に非対称性がみられるかどうかを集団のあいだの対立遺伝子の頻度の違いにもとづき推定するアルゴリズムとして∂a∂iがある7).ほかに,種の系統関係と個々の遺伝子の遺伝子系図のパターンの違いから遺伝子の流動を検出するABBA/BABAテストもよく利用されている8).
われわれヒトがひとりひとり身長や顔のかたちが違うように,ひとつの種のなかにはさまざまな表現型の変異が存在する.こういった変異を自然変異とよぶが,その多くはもともと個体ごとにゲノムのDNA配列が少しずつ異なることに起因する.リシークエンス解析の主要な目的のひとつは,このような種における表現型の変異をつかさどる遺伝変異をさぐることであり,その一般的な手法としてゲノムワイド関連解析(genome-wide association study:GWAS)がある.ゲノムワイド関連解析はヒトの遺伝疾患において関連遺伝子を同定する目的で以前から利用されてきたが,近年,シロイヌナズナやイネなど植物のゲノム研究においてもさかんに用いられるようになってきた.
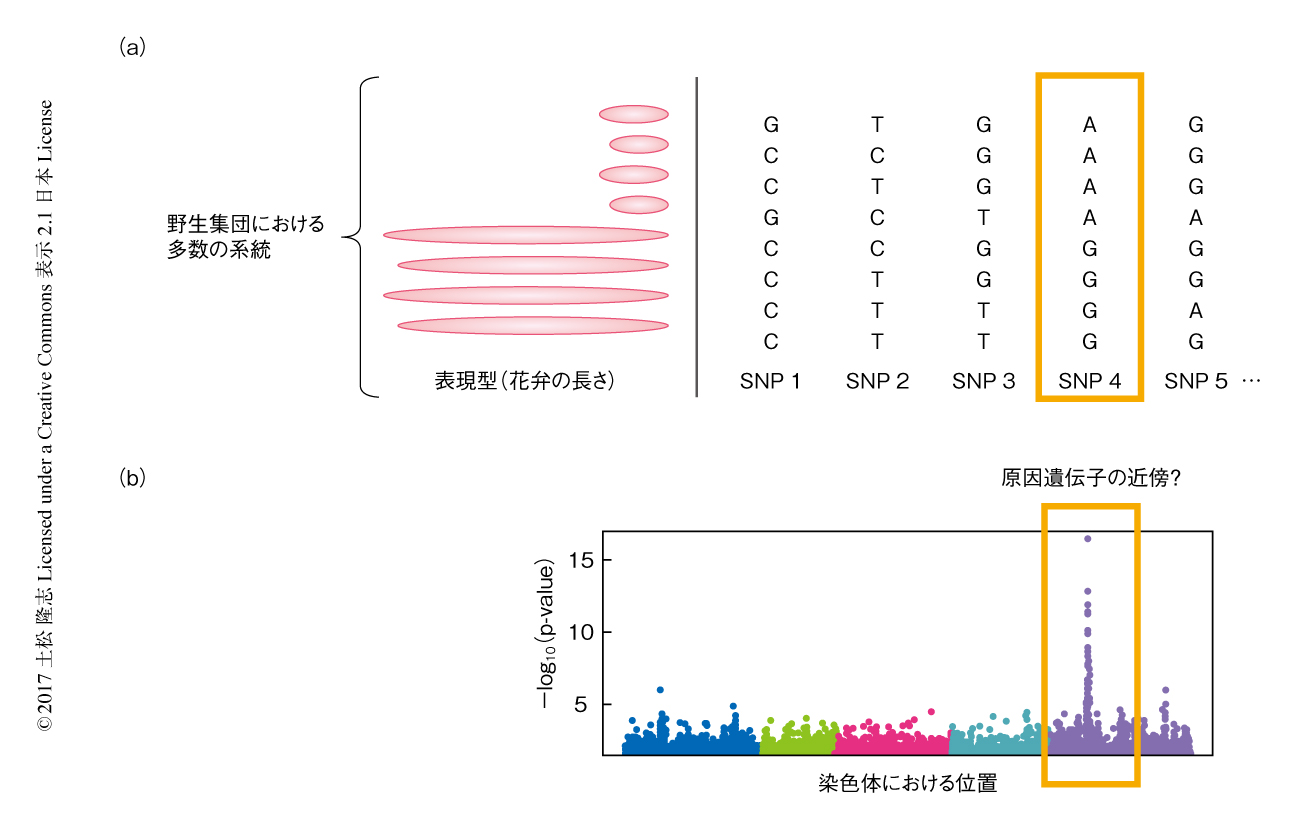
ゲノムワイド関連解析は集団における多数の個体について表現型のデータを取得し,表現型と遺伝子型との統計的な相関関係をゲノムの全体にわたり探索する手法である(図3a).ゲノムワイドな遺伝子型のデータセットとしてはSNPのマトリックスがまさに相当する.ゲノムワイド関連解析の典型的な結果は,ピークのさまがニューヨークのマンハッタン島のビル街のようであることからマンハッタンプロットとよばれる(図3b).表現型としては,疾患への感受性と抵抗性などの2値的な質的形質だけでなく,身長や体重などの量的形質も扱うことができる.
ゲノムワイド関連解析は,表現型の変異の原因となる多型と,そのごく近傍のSNPマーカーとのあいだに連鎖不平衡のあることを利用している.連鎖不平衡とは,SNPマーカーや表現型の変異の原因となる多型など2つの座位のあいだにみられる非独立(相関)関係のことである.連鎖不平衡は座位のあいだの組換えにより解消される.一般に,染色体における距離の近い座位のあいだの組換えはまれであるため,近傍の座位のあいだの連鎖不平衡は強くなる傾向がある.SNPマーカーが互いにどのくらい近くにあると連鎖不平衡がみられるかは種により大きく異なる.シロイヌナズナにおいてはおおむね10 kb以内であれば連鎖不平衡のみられることがわかっている.このことはすなわち,シロイヌナズナにおいてはゲノムワイド関連解析により理論上は10 kb程度の染色体の領域まで原因遺伝子座をしぼり込むことができることを意味する.
自然変異を担う遺伝子を同定する手法として,従来はF2集団などを用いた連鎖解析がよく用いられてきた.連鎖解析のなかでも,とくに量的形質を扱う際はQTL(quantitative trait loci,量的形質遺伝子座)解析とよばれる.量的形質の場合,表現型と遺伝子型との相関の統計学的な取り扱いがやや複雑になるものの,基本的な原理は共通する.
ゲノムワイド関連解析と連鎖解析にはそれぞれ長所と短所がある.ゲノムワイド関連解析の明らかにすぐれているところは,かけ合わせなどにより後代を作出する必要のない点である.これは,樹木など次世代をつくりだすのに多大な年月を要する種ではとくに重要なメリットだろう.もうひとつは,一般にゲノムワイド関連解析は連鎖解析よりもマッピングの解像度の高い点である.これは,マッピングに用いる集団における組換えの総量の違いに起因する.F2集団を用いた連鎖解析では両親のゲノムが組み換わるのはF1からF2の1世代のみである一方,ゲノムワイド関連解析が対象とする自然集団では世代ごとに何万年,何十万年と組換えをくり返しており,集団における組換えの数はF2集団よりもずっと多くなる.具体的な解像度としては,さきに述べたとおり,原因の突然変異と連鎖不平衡の関係にある染色体の領域(多くの種では,数百bp~数十kb)にまでしぼることができると考えられる.これだけ短い領域には数個の遺伝子しか含まれないことが多く,自然変異にかかわる遺伝子をゲノムワイド関連解析によりピンポイントに同定することも可能になってくる.
一般に,ゲノムワイド関連解析には連鎖解析よりずっと多くの遺伝マーカーが必要になる.これは,ゲノムワイド関連解析はSNPマーカーと原因遺伝子とのあいだの連鎖不平衡を利用したマッピングであるため,連鎖不平衡がみられる平均的な距離(種により異なるが,だいたい数百bp~数十kb)より十分に密な間隔でSNPマーカーが設計されている必要があるからである.もしSNPマーカーの密度が低すぎると,原因遺伝子と連鎖不平衡の関係にあるSNPマーカーがひとつも存在しないという事態が起こりうる.なお,シロイヌナズナにおいてゲノムワイド関連解析によく利用されてきたSNPマーカーのデータセットは約214,000万個のSNPからなる9).シロイヌナズナのゲノムのサイズは約130 Mbなので,平均して約600 bpに1個の間隔で遺伝マーカーが存在することになる.これは,シロイヌナズナにおいて連鎖不平衡のみられる平均的な距離である約10 kbよりずっと短い間隔である.一方,F2集団などを用いた連鎖解析においては一般にそこまで多くの遺伝マーカーは必要なく,せいぜい数百個のマーカーがあれば染色体におけるおおまかな位置を特定できる.これは,連鎖解析の場合,マッピングの律速は遺伝マーカーの数よりむしろ組換えの数であり,あまり遺伝マーカーを増やしても解像度は頭打ちになるということでもある.組換えの数を増やすには解析する個体数を増やす必要がある.
ゲノムワイド関連解析において重要な問題点として指摘されているのが,検出の際の偽陽性および偽陰性である.この場合,偽陽性とは実際には原因遺伝子ではないのに検出されてしまうことをさし,偽陰性とは実際には原因遺伝子なのに検出されないことをさす.
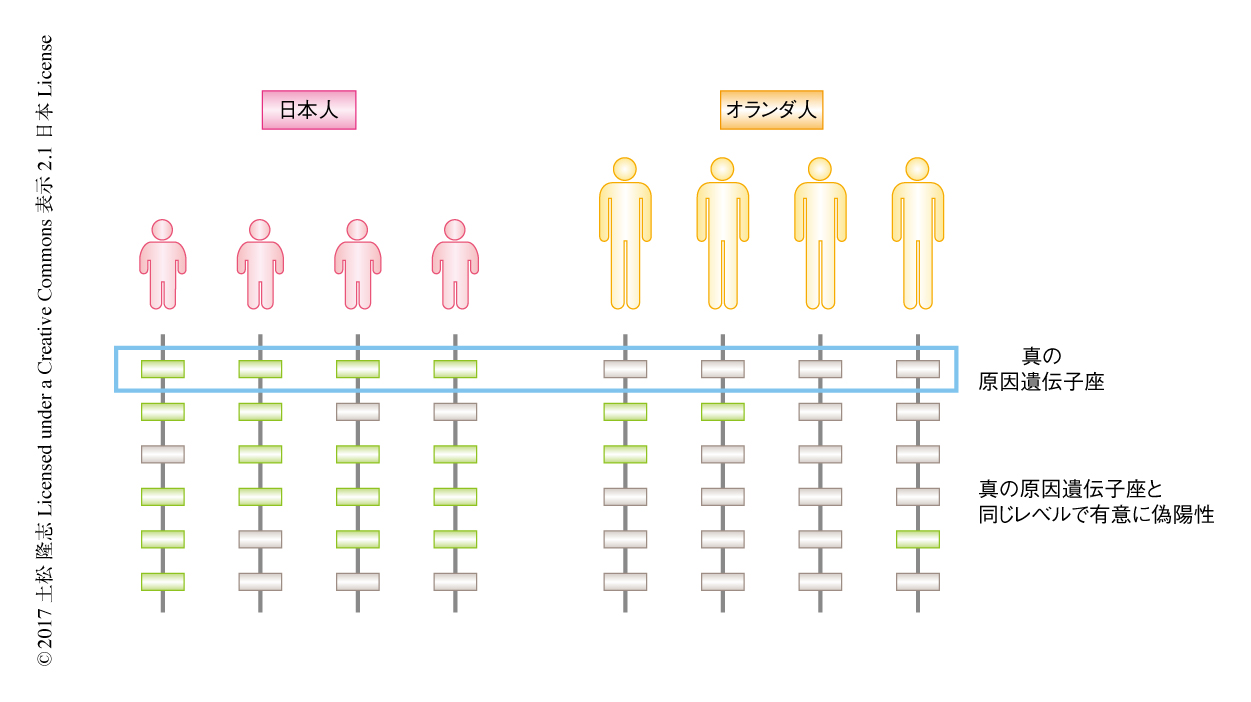
偽陽性の生じるおもな要因は,対象とする種や集団においてみられる集団構造である.種や集団の全体が遺伝的に均一に混ざりあっておらず分集団のような構造がみられると,分集団ごとにSNPの対立遺伝子の頻度が異なることが予想される.このとき,対象とする表現型の頻度も分集団ごとに異なると,ゲノムの多くのSNPが,実際には表現型と何の関係もないにもかかわらず,みかけ上の相関を示してしまうことがある.極端な例を考えてみるとわかりやすい.ヒトの身長にかかわる遺伝子を調べるため,日本人1000人とオランダ人1000人,合計2000人についてゲノムワイド関連解析を実施することを考える(図4).おおむね日本人は背が低くオランダ人は背が高いため,たとえば,日本人の多くがA,オランダ人の多くがTをもつようなSNPは,身長と強い相関を示すだろう.では,このSNPは本当に身長の変異に関与するといえるだろうか.もしかしたらそうかもしれないが,ほとんどの場合は違うだろう.日本人の集団とオランダ人の集団とはまったくランダムには混ざりあっていないため,それぞれの集団に固有の,そして,身長にはいっさい関係のないSNPがゲノムに大量に存在するのは自明なことである.この例は極端であり,日本人を1000人,オランダ人を1000人というサンプリングのしかたは明らかに不適切である.しかし,実際の自然集団には多かれ少なかれ集団構造があるため,このような効果をうまく除いてゲノムワイド関連解析を実施することが重要になる.ゲノムワイド関連解析の偽陽性を減らすために,これまで,さまざまな手法が提案されてきた.現在,よく使われているのは,線形混合モデルとよばれる統計的な手法による集団構造の補正である.いくつかのソフトウェアが提供されており,代表的なものとして,EMMAX 10) やGEMMA 11) がある.連鎖解析は基本的にこのような集団構造の影響をうけない.ゲノムワイド関連解析と連鎖解析の両方を実施し,どちらも同じ染色体の領域に検出された場合は,かなり信頼性が高いといえるだろう.
このような集団構造の効果による偽陽性にくわえ,ゲノムワイド関連解析には偽陰性の問題もある.偽陰性は,要は検出力の不足ということであり,その要因もさまざまである.たとえば,表現型にかかわる遺伝変異の集団における頻度がごく低いときや,その遺伝子が表現型におよぼす効果が小さいとき,あるいは,同じ表現型をもつ個体であっても別の遺伝子の変異が原因である場合などは,ゲノムワイド関連解析において検出されにくくなると考えられる.一般に,試料の数を増やすことにより検出力は上昇するものの,表現型の原因となる突然変異の頻度が極端に低いときなどは検出がむずかしいだろう.
偽陽性や偽陰性の問題はあるものの,これまでに,ゲノムワイド関連解析を用いて自然変異にかかわるさまざまな遺伝子が同定されてきた.シロイヌナズナにおいては,開花の時期や病害に対する抵抗姓などに関して,以前の遺伝学的な研究から機能が既知であった遺伝子がゲノムワイド関連解析により多くヒットしたほか12-14),根のメリステムのサイズの変異15) や塩耐性16) にかかわる遺伝子などについては,ゲノムワイド関連解析によりまったく新規の遺伝子が同定された.また最近,イネにおいてもゲノムワイド関連解析がさかんに実施されており,芒(のぎ)の長さ,開花,植物体の背丈などに関連する遺伝子が同定されている17,18)(文献18) は新着論文レビュー でも掲載).このように,ゲノムワイド関連解析は新規の遺伝子を探索するツールとしておもに利用されているが,一方で,自然集団に存在する遺伝変異と表現型との関係を直接に調べるものであるため,その解析自体が進化生物学的に重要な示唆をあたえうる.興味深い研究例をいくつか紹介する.
シロイヌナズナは北半球を中心に広い分布をもつため,その生育する気候も系統によりさまざまである.おのおのの系統は湿度や気温などその生育場所の気候に適応して進化してきたと考えられ,それぞれの集団においては有利な対立遺伝子が固定していることが予想される.そこで,おのおの系統の採集された場所の気候のさまざまなパラメーターそれ自体をその系統の表現型とみなし,気候のパラメーターについてゲノムワイド関連解析が実施された19).その結果,特定の1個の高いピークがみられたというより,比較的効果の小さいピークがゲノムの全体にわたり多数みつかった.それらのピークは,水分の欠乏への応答系や耐寒性にかかわる遺伝子など,環境応答に関連する遺伝子が有意に多い傾向があった.この結果をふまえ,フランス北部の都市リールにおいて圃場における実験が実施された.リールもシロイヌナズナの自然分布域にあるため,ゲノムワイド関連解析によりヒットしたおのおののSNPについて,リールの気候により適したほうの対立遺伝子を予測することができる.そして,147系統の野生系統を圃場にて栽培し,それぞれの適応度の指標として種子の生産数を測定したところ,リールの気候により適したほうの対立遺伝子をより多くもつ系統ほど,たしかに種子の生産数がより多くなった.この研究のように,ゲノムワイド関連解析による予測と圃場における検証をとおして,気候への適応は効果の小さい多数の遺伝子座の足し合わせにより起こったことが示された.
この研究においては生育場所の気候を表現型とみなしたが,ほかにも興味深い“表現型”を解析した例がある.圃場において196系統のシロイヌナズナを大量に栽培し,葉の表面にどのような微生物の群集がみられるかをメタゲノム解析により探索した20).おのおのの系統につき4個体ずつ解析したところ,どのような微生物がどれだけ存在するかは系統ごとにある程度の傾向のあることがわかった.この結果から,宿主であるシロイヌナズナの遺伝的な背景が,葉の表面に存在する微生物の群集に影響をおよぼすことが示唆された.そこで,微生物の群集の種の多様性をシロイヌナズナの表現型とみなしてゲノムワイド関連解析を実施したところ,トライコム(葉の表面の微細な毛)の形成や病害に対する抵抗姓にかかわる遺伝子などがヒットした.これは,いわばドーキンスの提案した“延長された表現型”であり,群集の組成がその宿主のもつ多数の遺伝子の遺伝子型により制約されることが示唆された.
このレビューにおいては,ひとつの種あるいは複数の近縁種の多数の個体についてSNPのデータセットを作成し,そこから集団の歴史の推定やゲノムワイド関連解析を実施する手法およびその適用例を紹介した.ここではふれなかったが,このような集団ゲノミクスのデータからほかにできることとして,おのおのの遺伝子座にはたらいてきた自然選択の検出がある.自然選択の解析とゲノムワイド関連解析とを組み合わせることにより,注目する表現型にいつどの程度の強さの自然選択がはたらいてきたのかを遺伝子のレベルから推定することも可能になる.
植物における大規模な集団ゲノミクスの研究は,研究者の人口や必要な情報の整備の状況などの理由から,依然としてシロイヌナズナやイネなどのモデル生物にかぎられるのが現状である.しかしながら,塩基配列決定技術の飛躍的な向上はいまも日進月歩につづいており,このような手法がいわゆる非モデル生物にも本格的に適用されるようになる日は近いだろう.さらなる研究の発展に期待したい.
略歴:2010年 東京大学大学院総合文化研究科博士課程 修了,スイスZurich大学 博士研究員,オーストリアGregor Mendel Institute博士研究員,東京大学大学院総合文化研究科 助教を経て,2016年より千葉大学大学院理学研究院 准教授.
研究テーマ:集団のゲノムデータを用いた植物の生殖システムの進化の理解.
研究室URL:https://tsuchimatsu.wordpress.com
© 2017 土松 隆志 Licensed under CC 表示 2.1 日本
(千葉大学大学院理学研究院 生物学研究部門植物進化ゲノミクス研究室)
email:土松隆志
領域融合レビュー, 6, e006 (2017) DOI: 10.7875/leading.author.6.e006
Takashi Tsuchimatsu: Population genomics reveals evolutionary processes of plant populations.
要 約
塩基配列決定技術の飛躍的な発展にともない,近年,全ゲノム情報にもとづく植物の進化の研究がさかんである.とくに,ひとつの種あるいは複数の近縁種にフォーカスして数十から数千におよぶ多数の試料をゲノム配列にもとづき解析する“集団ゲノミクス”の進展がめざましい.集団のゲノムデータを解析することにより,過去から現在にいたる個体数の変動や分化の過程などの歴史を推定できるほか,ゲノムワイド関連解析により自然変異にかかわる遺伝子を効率的に同定することも可能である.このレビューにおいては,モデル植物であるシロイヌナズナの事例を中心に,集団ゲノミクスの手法を解説する.
はじめに
生物のもつ全遺伝情報であるゲノムには,おもに2つの側面がある.1つ目は,ゲノムは生命活動に必要な情報のコードされた遺伝子の総体であり,さまざまな表現型がかたちづくられる“設計図”としての側面である.そして2つ目は,ゲノムは親から子へと脈々と受け継がれるものであり,生物の進化の痕跡がきざまれた“進化の履歴”としての側面である.現代の進化生物学における主要な課題のひとつは,ゲノムをこの2つの視点から詳細に解剖することにより,生物の集団にみられるさまざまな進化の過程を遺伝子のレベルから復元し予測することである.適応的な表現型はどの遺伝子のどの突然変異により生じてきたのだろうか.まったく新奇な表現型の進化には何個の遺伝子に変異が必要なのだろうか.ある生物種の集団は過去から現在にわたりどのように個体数が変動してきたのだろうか.近縁種どうしの種分化はいつ起こり,そののち,遺伝子の流動はどのくらい起こっているだろうか.塩基配列決定技術の向上にともない,ひとつの種あるいは近縁種における塩基配列の違いを全ゲノムにわたり容易かつ網羅的に把握できるようになったこともあり,近年,これらの問いに答える集団遺伝学の手法も飛躍的に発展している.
このレビューにおいておもにとりあげるのは,植物のひとつの種あるいは複数の近縁種にフォーカスして数十から数千におよぶ多数の試料をゲノム配列にもとづき解析する“集団ゲノミクス”である.はじめに,集団ゲノミクスにおいて扱うデータセットがどのようなものかを解説したうえで,個体数の動態など集団の歴史をどのように推定するか,また,表現型の変異をつかさどる遺伝子をどのように推定するか,という2つの視点から話を進める.
1.集団ゲノミクスに用いるデータセット
集団ゲノミクスの基本になるデータセットは,ひとつの種あるいは近縁種の数十から数千に及ぶ多数の個体についてゲノムを網羅する大量の遺伝マーカーのデータである.遺伝マーカーは,以前はSanger法などにより得られる断片的な塩基配列にもとづくものが多かったが,最近は次世代シークエンサーを利用し全ゲノム配列を決定するリシークエンス解析によるものが主流になりつつある.リシークエンス解析とは,すでにゲノム配列が解読されている種について,その既知のゲノム配列を参照配列として,次世代シークエンサーにより得られたほかの個体のゲノム配列に由来する短い塩基配列(ショートリード)をマッピングし,参照配列との違いのある部分を検出していく手法である(図1a).参照配列との違いとして得られるものには1塩基多型(single nucleotide polymorphism:SNP)や短い挿入および欠失(indel)などがあるが,もっとも頻繁にみられるSNPを遺伝マーカーとして用いることが多い.標準系統の参照配列に対しショートリードをマッピングし,ゲノムの全体にわたりSNPを抽出する.この作業を多数の個体について行うことにより,SNPの巨大なマトリックスを作成することができる(図1b).これが,さまざまな集団ゲノミクス解析の基礎となる重要なデータセットである.
このSNPのデータセットを用いることにより,たとえば,それぞれの個体が互いにどのくらい近縁かを簡単に計算することができる.ある2つの系統を抽出し,SNPを数えてゲノムの全長で割り算をすると,ゲノムにおいて何%の塩基に置換がみられるかがもとめられる.この値はより近縁であれば小さく,より遠縁であれば大きくなる.すべての個体の組合せについてこの計算を行い平均した値は塩基多様度とよばれ,その種や集団の遺伝的な多様性の基本的な指標になる.塩基多様度は,たとえば,シロイヌナズナで約0.5%,ヒトでは約0.1%であることが知られている.
このような個体のあいだの近縁度の全体像をおおまかに把握するため,ゲノムの類似度を主成分分析などの手法により2次元の平面に投影することがよく行われる.たとえば,ヨーロッパにおけるヒトの集団の遺伝的な類縁度を約3000人,500,568遺伝マーカーの多型データを用いて解析し,主成分分析を用いて可視化した研究がある1).遺伝的な類縁度は集団のあいだの地理的な関係とおおむね対応しており,ヒトの交配や移動が地理的に限定されていることが示唆される.
主成分分析にくわえて,クラスタリング解析も一般的である2).STRUCTUREやその派生のソフトウェアは,集団の全体がN個の分集団に分かれると仮定したとき,おのおのの個体がX番目に属する確率をもとめるものである.もちろん,厳密にN個の分集団に分かれるわけではなく,特定の分集団への帰属がはっきりしない個体もみられることが多い.それでも,対象とする種がおおむね何個の分集団に分かれるのか,この解析からおおまかな目安をつけることは可能である.このように,種や集団が遺伝的に均一に混ざりあっておらず分集団のような構造がみられるとき,これを集団構造とよぶ.主成分分析やクラスタリング解析をとおして,対象とする種の集団構造のパターンを把握することができる.
2.集団の進化の歴史の推定
主成分分析やクラスタリング解析などから明らかにされた集団構造は,その種の進化の歴史を反映している.たとえば,その種が長期にわたり2つの分集団に分かれていたなら,クラスタリング解析により2つのクラスターにはっきり分かれることが予想される.
集団ゲノミクスのデータを用いることにより,集団構造を把握するだけでなく,その背景にある集団の進化の歴史を定量的に推定することができる.ここで,集団の歴史とは具体的に,過去における個体数の増減,種や集団の分化,遺伝子の流動の過程などのことをさす.このような推定のベースにあるのは,コアレセント理論(coalescent theory)とよばれる比較的新しい集団遺伝学の理論である.コアレセント理論とは,集団から得られたDNA配列の系譜を遺伝子系図(gene genealogy)として解析することにより,さまざまな進化的なパラメーターを定量的に推定する集団遺伝学の考え方のひとつである.コアレセント(coalescent)という語は“合着”あるいは“合祖”と訳され,集団のある2つのDNA配列が世代をさかのぼり共通の祖先にいたることをさす(図2).
コアレセント理論の核となるコンセプトは,得られたDNA配列の合着が何世代まえにさかのぼるか理論的な期待値がもとめられることである.そして,合着が何世代まえにどれだけ起こったかは,個体数の変動,自然選択,集団構造などの影響をうけて変化する.つまり逆に考えれば,試料として得られたDNA配列の合着がいつどれだけ起こったかを調べれば,過去の集団における個体数の変動などを推定できることになる.おおまかには,集団のサイズが大きいときには合着は相対的に少なく,集団のサイズが小さいときにはより多くの合着が起こる.これは,あるタイミングで個体数が急に1個体に減少した状況を考えるとわかりやすい.すべての試料は最終的に必ずそこで合着するはずである.また,過去のある時期に集団が2つの分集団に分かれていたとすると,その期間の分集団のあいだにおいて合着は少ないことが期待される.
最近,このようなコアレセント理論による個体数の動態の推定を,多数の個体のゲノムワイドな遺伝子多型のデータにもとづき行うアルゴリズムが提案された.MSMC(multiple sequentially Markovian coalescent)とよばれるこの手法は3),2倍体の1個体のゲノム配列から過去の個体数の動態を推定するアルゴリズムPSMC(pairwise sequentially Markovian coalescent)を発展させたものである4).
ここでは,MSMC解析をシロイヌナズナのデータに適用した例を紹介する.シロイヌナズナはおもに中央アジア,ヨーロッパ,北アフリカ,北アメリカに生育する1年草であり,近年,“1001ゲノムプロジェクト”とよばれる1135に及ぶ野生系統のリシークエンス解析が実施されるなど,集団ゲノミクスのモデルとして注目されている5).1001ゲノムプロジェクトのリシークエンスデータをもとにSNPのマトリックスを作成し,すべての系統の組合せについて塩基置換率を計算したところ,おおむね,ピークを約0.005(0.5%)とするきれいな山形になったが,ピークより値の大きい側の裾野にかたよってふくらみがみられた.これは,ほかの大多数の系統とは遠縁な個体がいくつかまぎれていることによると考えられた.調べてみると,この遠縁な系統(ここでは,“遺存系統”とよぶ)は26系統あり,その多くは地中海地方に由来するものだった.MSMC解析を実施したところ,遺存系統と残りの大多数の系統とのあいだにおいては,最終氷期のはじまった7万年前ごろから最終氷期ののちの現在にいたるまで,ほとんど合着がみられないことがわかった5).この結果から,両者のあいだで最終氷期ののちには遺伝的な交流はほとんど起こっていないことが示された.最終氷期におけるシロイヌナズナの分布は南の地中海地方に制限され,いくつかの氷河退避地に集団が分かれていたと考えられている.最終氷期ののち,氷河退避地からふたたび分布が拡大した際に,おそらくはひとつの氷河退避地に由来する系統が圧倒的に頻度を高め,それが現在の多数派の系統となったと予想された.一方,遺伝的に分化した少数派の遺存系統は,いわば氷河期の生き残りとでもよぶべき系統であり,生育地の特性などの表現型に多数派の系統とは異なる特徴がみられると報告されている.
MSMC解析はシロイヌナズナだけでなく,最近,トウモロコシの集団ゲノミクスにも適用されている6).その結果,トウモロコシは近縁の野生種であるテオシンテから1万~1.5万年前に分岐し,その際,集団のサイズが約5%に減少する非常に強いボトルネック効果(びん首効果)のはたらいたことがわかった.そののち,集団のサイズは急速に増加し,現在の栽培トウモロコシの集団ができあがったと推定されている.
コアレセント理論とゲノムデータにもとづき集団の歴史を推定する手法は,MSMC解析のほかにもさまざまな手法が提案されている.たとえば,2つの集団のあいだで遺伝子の流動がどのくらい起こっているか,とくに,その遺伝子の流動に非対称性がみられるかどうかを集団のあいだの対立遺伝子の頻度の違いにもとづき推定するアルゴリズムとして∂a∂iがある7).ほかに,種の系統関係と個々の遺伝子の遺伝子系図のパターンの違いから遺伝子の流動を検出するABBA/BABAテストもよく利用されている8).
3.ゲノムワイド関連解析の原理
われわれヒトがひとりひとり身長や顔のかたちが違うように,ひとつの種のなかにはさまざまな表現型の変異が存在する.こういった変異を自然変異とよぶが,その多くはもともと個体ごとにゲノムのDNA配列が少しずつ異なることに起因する.リシークエンス解析の主要な目的のひとつは,このような種における表現型の変異をつかさどる遺伝変異をさぐることであり,その一般的な手法としてゲノムワイド関連解析(genome-wide association study:GWAS)がある.ゲノムワイド関連解析はヒトの遺伝疾患において関連遺伝子を同定する目的で以前から利用されてきたが,近年,シロイヌナズナやイネなど植物のゲノム研究においてもさかんに用いられるようになってきた.
ゲノムワイド関連解析は集団における多数の個体について表現型のデータを取得し,表現型と遺伝子型との統計的な相関関係をゲノムの全体にわたり探索する手法である(図3a).ゲノムワイドな遺伝子型のデータセットとしてはSNPのマトリックスがまさに相当する.ゲノムワイド関連解析の典型的な結果は,ピークのさまがニューヨークのマンハッタン島のビル街のようであることからマンハッタンプロットとよばれる(図3b).表現型としては,疾患への感受性と抵抗性などの2値的な質的形質だけでなく,身長や体重などの量的形質も扱うことができる.
ゲノムワイド関連解析は,表現型の変異の原因となる多型と,そのごく近傍のSNPマーカーとのあいだに連鎖不平衡のあることを利用している.連鎖不平衡とは,SNPマーカーや表現型の変異の原因となる多型など2つの座位のあいだにみられる非独立(相関)関係のことである.連鎖不平衡は座位のあいだの組換えにより解消される.一般に,染色体における距離の近い座位のあいだの組換えはまれであるため,近傍の座位のあいだの連鎖不平衡は強くなる傾向がある.SNPマーカーが互いにどのくらい近くにあると連鎖不平衡がみられるかは種により大きく異なる.シロイヌナズナにおいてはおおむね10 kb以内であれば連鎖不平衡のみられることがわかっている.このことはすなわち,シロイヌナズナにおいてはゲノムワイド関連解析により理論上は10 kb程度の染色体の領域まで原因遺伝子座をしぼり込むことができることを意味する.
4.ゲノムワイド関連解析の長所と短所
自然変異を担う遺伝子を同定する手法として,従来はF2集団などを用いた連鎖解析がよく用いられてきた.連鎖解析のなかでも,とくに量的形質を扱う際はQTL(quantitative trait loci,量的形質遺伝子座)解析とよばれる.量的形質の場合,表現型と遺伝子型との相関の統計学的な取り扱いがやや複雑になるものの,基本的な原理は共通する.
ゲノムワイド関連解析と連鎖解析にはそれぞれ長所と短所がある.ゲノムワイド関連解析の明らかにすぐれているところは,かけ合わせなどにより後代を作出する必要のない点である.これは,樹木など次世代をつくりだすのに多大な年月を要する種ではとくに重要なメリットだろう.もうひとつは,一般にゲノムワイド関連解析は連鎖解析よりもマッピングの解像度の高い点である.これは,マッピングに用いる集団における組換えの総量の違いに起因する.F2集団を用いた連鎖解析では両親のゲノムが組み換わるのはF1からF2の1世代のみである一方,ゲノムワイド関連解析が対象とする自然集団では世代ごとに何万年,何十万年と組換えをくり返しており,集団における組換えの数はF2集団よりもずっと多くなる.具体的な解像度としては,さきに述べたとおり,原因の突然変異と連鎖不平衡の関係にある染色体の領域(多くの種では,数百bp~数十kb)にまでしぼることができると考えられる.これだけ短い領域には数個の遺伝子しか含まれないことが多く,自然変異にかかわる遺伝子をゲノムワイド関連解析によりピンポイントに同定することも可能になってくる.
一般に,ゲノムワイド関連解析には連鎖解析よりずっと多くの遺伝マーカーが必要になる.これは,ゲノムワイド関連解析はSNPマーカーと原因遺伝子とのあいだの連鎖不平衡を利用したマッピングであるため,連鎖不平衡がみられる平均的な距離(種により異なるが,だいたい数百bp~数十kb)より十分に密な間隔でSNPマーカーが設計されている必要があるからである.もしSNPマーカーの密度が低すぎると,原因遺伝子と連鎖不平衡の関係にあるSNPマーカーがひとつも存在しないという事態が起こりうる.なお,シロイヌナズナにおいてゲノムワイド関連解析によく利用されてきたSNPマーカーのデータセットは約214,000万個のSNPからなる9).シロイヌナズナのゲノムのサイズは約130 Mbなので,平均して約600 bpに1個の間隔で遺伝マーカーが存在することになる.これは,シロイヌナズナにおいて連鎖不平衡のみられる平均的な距離である約10 kbよりずっと短い間隔である.一方,F2集団などを用いた連鎖解析においては一般にそこまで多くの遺伝マーカーは必要なく,せいぜい数百個のマーカーがあれば染色体におけるおおまかな位置を特定できる.これは,連鎖解析の場合,マッピングの律速は遺伝マーカーの数よりむしろ組換えの数であり,あまり遺伝マーカーを増やしても解像度は頭打ちになるということでもある.組換えの数を増やすには解析する個体数を増やす必要がある.
ゲノムワイド関連解析において重要な問題点として指摘されているのが,検出の際の偽陽性および偽陰性である.この場合,偽陽性とは実際には原因遺伝子ではないのに検出されてしまうことをさし,偽陰性とは実際には原因遺伝子なのに検出されないことをさす.
偽陽性の生じるおもな要因は,対象とする種や集団においてみられる集団構造である.種や集団の全体が遺伝的に均一に混ざりあっておらず分集団のような構造がみられると,分集団ごとにSNPの対立遺伝子の頻度が異なることが予想される.このとき,対象とする表現型の頻度も分集団ごとに異なると,ゲノムの多くのSNPが,実際には表現型と何の関係もないにもかかわらず,みかけ上の相関を示してしまうことがある.極端な例を考えてみるとわかりやすい.ヒトの身長にかかわる遺伝子を調べるため,日本人1000人とオランダ人1000人,合計2000人についてゲノムワイド関連解析を実施することを考える(図4).おおむね日本人は背が低くオランダ人は背が高いため,たとえば,日本人の多くがA,オランダ人の多くがTをもつようなSNPは,身長と強い相関を示すだろう.では,このSNPは本当に身長の変異に関与するといえるだろうか.もしかしたらそうかもしれないが,ほとんどの場合は違うだろう.日本人の集団とオランダ人の集団とはまったくランダムには混ざりあっていないため,それぞれの集団に固有の,そして,身長にはいっさい関係のないSNPがゲノムに大量に存在するのは自明なことである.この例は極端であり,日本人を1000人,オランダ人を1000人というサンプリングのしかたは明らかに不適切である.しかし,実際の自然集団には多かれ少なかれ集団構造があるため,このような効果をうまく除いてゲノムワイド関連解析を実施することが重要になる.ゲノムワイド関連解析の偽陽性を減らすために,これまで,さまざまな手法が提案されてきた.現在,よく使われているのは,線形混合モデルとよばれる統計的な手法による集団構造の補正である.いくつかのソフトウェアが提供されており,代表的なものとして,EMMAX 10) やGEMMA 11) がある.連鎖解析は基本的にこのような集団構造の影響をうけない.ゲノムワイド関連解析と連鎖解析の両方を実施し,どちらも同じ染色体の領域に検出された場合は,かなり信頼性が高いといえるだろう.
このような集団構造の効果による偽陽性にくわえ,ゲノムワイド関連解析には偽陰性の問題もある.偽陰性は,要は検出力の不足ということであり,その要因もさまざまである.たとえば,表現型にかかわる遺伝変異の集団における頻度がごく低いときや,その遺伝子が表現型におよぼす効果が小さいとき,あるいは,同じ表現型をもつ個体であっても別の遺伝子の変異が原因である場合などは,ゲノムワイド関連解析において検出されにくくなると考えられる.一般に,試料の数を増やすことにより検出力は上昇するものの,表現型の原因となる突然変異の頻度が極端に低いときなどは検出がむずかしいだろう.
5.植物におけるゲノムワイド関連解析の研究例
偽陽性や偽陰性の問題はあるものの,これまでに,ゲノムワイド関連解析を用いて自然変異にかかわるさまざまな遺伝子が同定されてきた.シロイヌナズナにおいては,開花の時期や病害に対する抵抗姓などに関して,以前の遺伝学的な研究から機能が既知であった遺伝子がゲノムワイド関連解析により多くヒットしたほか12-14),根のメリステムのサイズの変異15) や塩耐性16) にかかわる遺伝子などについては,ゲノムワイド関連解析によりまったく新規の遺伝子が同定された.また最近,イネにおいてもゲノムワイド関連解析がさかんに実施されており,芒(のぎ)の長さ,開花,植物体の背丈などに関連する遺伝子が同定されている17,18)(文献18) は新着論文レビュー でも掲載).このように,ゲノムワイド関連解析は新規の遺伝子を探索するツールとしておもに利用されているが,一方で,自然集団に存在する遺伝変異と表現型との関係を直接に調べるものであるため,その解析自体が進化生物学的に重要な示唆をあたえうる.興味深い研究例をいくつか紹介する.
シロイヌナズナは北半球を中心に広い分布をもつため,その生育する気候も系統によりさまざまである.おのおのの系統は湿度や気温などその生育場所の気候に適応して進化してきたと考えられ,それぞれの集団においては有利な対立遺伝子が固定していることが予想される.そこで,おのおの系統の採集された場所の気候のさまざまなパラメーターそれ自体をその系統の表現型とみなし,気候のパラメーターについてゲノムワイド関連解析が実施された19).その結果,特定の1個の高いピークがみられたというより,比較的効果の小さいピークがゲノムの全体にわたり多数みつかった.それらのピークは,水分の欠乏への応答系や耐寒性にかかわる遺伝子など,環境応答に関連する遺伝子が有意に多い傾向があった.この結果をふまえ,フランス北部の都市リールにおいて圃場における実験が実施された.リールもシロイヌナズナの自然分布域にあるため,ゲノムワイド関連解析によりヒットしたおのおののSNPについて,リールの気候により適したほうの対立遺伝子を予測することができる.そして,147系統の野生系統を圃場にて栽培し,それぞれの適応度の指標として種子の生産数を測定したところ,リールの気候により適したほうの対立遺伝子をより多くもつ系統ほど,たしかに種子の生産数がより多くなった.この研究のように,ゲノムワイド関連解析による予測と圃場における検証をとおして,気候への適応は効果の小さい多数の遺伝子座の足し合わせにより起こったことが示された.
この研究においては生育場所の気候を表現型とみなしたが,ほかにも興味深い“表現型”を解析した例がある.圃場において196系統のシロイヌナズナを大量に栽培し,葉の表面にどのような微生物の群集がみられるかをメタゲノム解析により探索した20).おのおのの系統につき4個体ずつ解析したところ,どのような微生物がどれだけ存在するかは系統ごとにある程度の傾向のあることがわかった.この結果から,宿主であるシロイヌナズナの遺伝的な背景が,葉の表面に存在する微生物の群集に影響をおよぼすことが示唆された.そこで,微生物の群集の種の多様性をシロイヌナズナの表現型とみなしてゲノムワイド関連解析を実施したところ,トライコム(葉の表面の微細な毛)の形成や病害に対する抵抗姓にかかわる遺伝子などがヒットした.これは,いわばドーキンスの提案した“延長された表現型”であり,群集の組成がその宿主のもつ多数の遺伝子の遺伝子型により制約されることが示唆された.
おわりに
このレビューにおいては,ひとつの種あるいは複数の近縁種の多数の個体についてSNPのデータセットを作成し,そこから集団の歴史の推定やゲノムワイド関連解析を実施する手法およびその適用例を紹介した.ここではふれなかったが,このような集団ゲノミクスのデータからほかにできることとして,おのおのの遺伝子座にはたらいてきた自然選択の検出がある.自然選択の解析とゲノムワイド関連解析とを組み合わせることにより,注目する表現型にいつどの程度の強さの自然選択がはたらいてきたのかを遺伝子のレベルから推定することも可能になる.
植物における大規模な集団ゲノミクスの研究は,研究者の人口や必要な情報の整備の状況などの理由から,依然としてシロイヌナズナやイネなどのモデル生物にかぎられるのが現状である.しかしながら,塩基配列決定技術の飛躍的な向上はいまも日進月歩につづいており,このような手法がいわゆる非モデル生物にも本格的に適用されるようになる日は近いだろう.さらなる研究の発展に期待したい.
文 献
- Novembre, J., Johnson, T., Bryc, K. et al.: Genes mirror geography within Europe. Nature, 456, 98-101 (2008)[PubMed]
- Pritchard, J. K., Stephens, M. & Donnelly, P.: Inference of population structure using multilocus genotype data. Genetics, 155, 945-959 (2000)[PubMed]
- Schiffels, S. & Durbin, R.: Inferring human population size and separation history from multiple genome sequences. Nat. Genet., 46, 919-925 (2014)[PubMed]
- Li, H. & Durbin, R.: Inference of human population history from individual whole-genome sequences. Nature, 475, 493-496 (2011)[PubMed]
- 1001 Genomes Consortium: 1,135 Genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell, 166, 481-491 (2016)[PubMed]
- Beissinger, T. M., Wang, L., Crosby, K. et al.: Recent demography drives changes in linked selection across the maize genome. Nat. Plants, 2, 16084 (2016)[PubMed]
- Gutenkunst, R. N., Hernandez, R. D., Williamson, S. H. et al.: Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet., 5, e1000695 (2009)[PubMed]
- Durand, E. Y., Patterson, N., Reich, D. et al.: Testing for ancient admixture between closely related populations. Mol. Biol. Evol., 28, 2239-2252 (2011)[PubMed]
- Horton, M. W., Hancock, A. M., Huang, Y. S. et al.: Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat. Genet., 44, 212-216 (2012)[PubMed]
- Kang, H. M., Sul, J. H., Service, S. K. et al.: Variance component model to account for sample structure in genome-wide association studies. Nat. Genet., 42, 348-354 (2010)[PubMed]
- Zhou, X. & Stephens, M.: Genome-wide efficient mixed model analysis for association studies. Nat. Genet., 44, 821-824 (2012)[PubMed]
- Atwell, S., Huang, Y. S., Vilhjalmsson, B. J. et al.: Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature, 465, 627-631 (2010)[PubMed]
- Baxter, I., Brazelton, J. N., Yu, D. et al.: A coastal cline in sodium accumulation in Arabidopsis thaliana is driven by natural variation of the sodium transporter AtHKT1;1. PLoS Genet., 6, e1001193 (2010)[PubMed]
- Brachi, B., Faure, N., Horton, M. et al.: Linkage and association mapping of Arabidopsis thaliana flowering time in nature. PLoS Genet., 6, e1000940 (2010)[PubMed]
- Meijon, M., Satbhai, S. B., Tsuchimatsu, T. et al.: Genome-wide association study using cellular traits identifies a new regulator of root development in Arabidopsis. Nat. Genet., 46, 77-81 (2014)[PubMed]
- Ariga, H., Katori, T., Tsuchimatsu, T. et al.: NLR locus-mediated trade-off between abiotic and biotic stress adaptation in Arabidopsis. Nat. Plants, 3, 17072 (2017)[PubMed]
- Huang X., Wei, X., Sang, T. et al.: Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet., 42, 961-967 (2010)[PubMed]
- Yano, K., Yamamoto, E., Aya, K. et al.: Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet., 48, 927-934 (2016)[PubMed] [新着論文レビュー]
- Hancock, A. M., Brachi, B., Faure, N. et al.: Adaptation to climate across the Arabidopsis thaliana genome. Science, 334, 83-86 (2011)[PubMed]
- Horton, M. W., Bodenhausen, N., Beilsmith, K. et al.: Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nat. Commun., 5, 5320 (2014)[PubMed]
著者プロフィール
略歴:2010年 東京大学大学院総合文化研究科博士課程 修了,スイスZurich大学 博士研究員,オーストリアGregor Mendel Institute博士研究員,東京大学大学院総合文化研究科 助教を経て,2016年より千葉大学大学院理学研究院 准教授.
研究テーマ:集団のゲノムデータを用いた植物の生殖システムの進化の理解.
研究室URL:https://tsuchimatsu.wordpress.com
© 2017 土松 隆志 Licensed under CC 表示 2.1 日本
